黏多糖贮积症是由于人体细胞的溶酶体内降解黏多糖的水解酶发生突变导致其活性丧失,黏多糖不能被降解代谢,最终贮积在体内而发生的疾病。该病是溶酶体贮积病中非常重要的一类,可分为Ⅰ,Ⅱ,Ⅲ,Ⅳ,Ⅵ,Ⅶ,Ⅸ型等7种型,其中Ⅲ又分为ⅢA,ⅢB,ⅢC,ⅢD四个亚型,Ⅳ型分为ⅣA和ⅣB亚型,虽然各型致病基因和临床表现有差异,但由于贮积的底物都是黏多糖而被统称为黏多糖贮积症。
粘多糖贮积症_黏多糖贮积症 -基本简介
多发性骨发育障碍
黏多糖贮积症(mucopolysaccharidosis,MPS)是一组溶酶体累积病,是由于溶酶体水解酶缺陷,造成酸性黏多糖(葡糖氨基聚糖)降解受阻黏多糖在体内积聚而引起一系列临床症状。黏多糖是结缔组织间的主要成分,包括透明质酸硫酸软骨素、硫酸皮肤素、硫酸类肝素和硫酸角质素,这些多糖都是直链杂多糖,可同时与一条蛋白质肽链结合,聚合成更大的分子。正常溶酶体中含有许多种糖苷酶,其中有10种参与葡糖氨基聚糖链的降解过程它们中任何一种糖苷酶的缺陷都会造成葡糖氨基聚糖链分解障碍而在溶酶体内积聚,并自尿中排出。
黏多糖贮积症患者由于过多的黏多糖贮积于骨、软骨等组织或器官内从而影响到这些组织或器官的正常发育,多余的黏多糖从尿中排出,发生一系列的临床症状和影像学表现。黏多糖贮积症属先天性或原发性代谢异常综合征。
根据尿糖中所含酸性黏多糖的种类,相关个别酶缺乏和活性低下的种类以临床表现和影像学表现的不同,将黏多糖贮积症分为7大类型,每一型又分为2~4个亚型其中黏多糖贮积症ⅠⅣ型最为常见且较具特征性,而尤以Ⅰ型最典型,为黏多糖贮积症的原型。
黏多糖贮积症在北美和欧洲的总发病率约为1/25000。国外已经有针对黏多糖贮积症某些亚型的酶替代治疗药物,但这些药物还未在国内上市,即使上市,每年高达百万元的费用且需要终身用药,若无医疗保险支持大部分家庭也无力承担。该群体也被称为“黏宝宝”。
粘多糖贮积症_黏多糖贮积症 -发病原因
β-半乳糖苷酶由于黏多糖增多症是一类非常罕见的疾病尚缺乏有关本症患病率或发病率方面的确切资料。北美和欧洲各型黏多糖增多症的总发病率约为1∶2.5万。
病因:
1.黏多糖贮积症Ⅰ型(Hurler综合征)为带染色体隐性遗传疾病,是由于α-L-艾杜糖酶(α-L-iduronidase)缺乏所致,可分为3个亚型:
(1)Hurler综合征:即MPSIH型。
(2)Scheie综合征即MPS-IS型,亦即7大类中原Ⅴ型(MPS-Ⅴ型)
(3)Hurler-Sheie综合征其改变介于前两型之间。
2.黏多糖贮积症Ⅱ型(Hunter综合征)为伴性(X)连锁遗传性疾病,仅见于男性,由于体内缺乏艾杜糖醛酸硫酸酶而患病临床表现与X线检查同MPS-1,但其临床进展慢于前者,临床表现轻于前者,该型根据临床表现轻重,又分2个亚型是:①MPSⅡA又称重症型;②MPSⅡB又称轻症型。
3.黏多糖贮积症Ⅲ型(Sanfilippo综合征)旧称营养不良性智力发育不全(polydystrophicoligophrenia)为常染色体隐性遗传性疾病,体内多种酶缺乏特征性临床表现为进行性智力低下,其他如面貌、身材改变,严重程度不一。根据缺乏酶的不同和临床表现的差异等,又可分4个亚型,即MPSⅢA、MPSⅢBMPSⅢC和MPSⅢD。
4.黏多糖贮积症Ⅳ型(Morquio综合征)为较多见的黏多糖贮积症,属常染色体隐性遗传。临床表现较独特。本型分2个亚型:
(1)MPSⅣA型,其相关缺乏酶为N-乙酰-半乳糖氨糖-6-硫酸盐硫酸酯酶(N-acetyl-galactosamine-6-sulfatesulfatase)。
(2)MPSⅣB型,缺乏酶为β-半乳糖苷酶(bata-galactosidase)。该2个亚型,临床表现严重程度上可差异较大,通常A型病情较严重。
5.黏多糖贮积症Ⅴ型现认为该型即为黏多糖贮积症Ⅰ型的Seheie型,与Hrular综合征不同之处表现为无严重的角膜混浊,且混浊为周边性患者智力正常,身材正常或稍矮,寿命基本正常,但有多毛,关节强直。背柱、头颅X线示仅有轻微改变。
6.黏多糖贮积症Ⅵ型(Maroteaux-Lamy综合征)或称芳基硫酸酯酶B缺乏症(anglsulfataseBdeficiency)。为常染色体隐性遗传疾病,缺乏酶即为芳基硫酸酯酶。本型与Hurler综合征基本相似但智力正常,与Hurler不同者为部分病人尚有骨骺,尤其是股骨头骺后缺血坏死样改变可存在该症预后较MPSⅠ综合征寿命长与Hurler鉴别诊断主要根据寿限较长智力基本正常,及骨骺可存在Hurler尿中硫酸皮肤素及硫酸肝素均增多而Ⅵ型仅后者增多。在缺乏酶方面Ⅰ型缺α-L-艾杜糖醛酶,Ⅵ型缺芳基硫酸酯酶B。
7.黏多糖贮积症Ⅶ型(Sly综合征)为常染色体隐性遗传病,极罕见,患者缺乏β-葡萄糖醛酸酶(β-glucuronidase),患者婴儿期即见身材矮小,智力迟钝鸡胸,背柱侧弯等本症分重症和轻症2个亚型,前者发病早,并有关节挛缩,后者发病晚,常有股骨头缺血坏死样改变。
粘多糖贮积症_黏多糖贮积症 -发病机制
X染色体除MPSⅡ型呈X连锁隐性遗传外,其余各型的遗传方式均为常染色体隐性遗传有缺陷的基因定位于常染色体上,且只有纯合子基因型才会发病在父母均为杂合子的子女中其基因突变与正常的几率均为25%,其余50%均为杂合子基因携带者由于MPSⅡ型的缺陷基因位于性染色体X上因此只有男性才患病,女性均为基因携带者,子代中男性患病以及女性成为携带者的几率各为50%
已证实的基因突变种类繁多,且不同人群之间的差异较大。
黏多糖包括4-硫酸软骨素、6-硫酸软骨素、硫酸软骨素、硫酸类肝素、硫酸角质素、肝素及透明质酸等成分,为角膜、软骨骨骼、皮肤、筋膜、心瓣膜和血管结缔组织的结构成分。MPSⅠ型的α-艾杜糖醛酸酶缺乏、MPSⅡ型的艾杜糖醛酸硫酸酯酶缺乏以及MPSⅦ型的β-葡萄糖醛酸酶缺乏,均导致硫酸软骨素和硫酸类肝素的降解受阻。MPSⅢ型的各种酶缺乏均可引起硫酸类肝素的降解障碍。MPSⅣ型的β-半乳糖苷酶缺乏主要影响硫酸角质素的降解。MPSⅥ型的酰基硫酸酯酶B缺乏主要使硫酸软骨素的降解受阻。不能降解的各种黏多糖成分在体内大量积蓄,并沉积于上述各组织中,引起器官损害及功能障碍。同时,过多的黏多糖可从尿液中不断排出。
粘多糖贮积症_黏多糖贮积症 -临床表现
大多数患儿出生时正常,1岁以内的生长与发育亦基本正常。发病年龄因黏多糖增多症的类型不同而各有差异。初发症状多为耳部感染、流涕和感冒等。
虽然各型黏多糖增多症的病程进展与病情严重程度差异较大,但患儿在临床表现方面具有某些共同的特征如:身材矮小特殊面容及骨骼系统异常等。多数患儿都有关节改变和活动受限。多器官受累见于所有的患儿。部分患儿有角膜混浊,并可因此而导致视力障碍甚至失明。肝脾肿大以及心血管受累较为常见。部分患儿可有智力发育进行性迟缓,脐疝和腹股沟疝,生长缓慢,脑积水,皮肤增厚,毛发增多,慢性流涕耳部反复感染,并可致听力损害等。
表1各型黏多糖贮积症的特征(表1)。
1.黏多糖贮积症Ⅰ型虽然黏多糖贮积症Ⅰ有3种亚型但均为同一种酶缺乏,只是酶缺乏的程度不同而已。其中以Hurler综合征较常见,临床表现最为严重,Scheie综合征的症状出现时间较晚,病情最轻,而Hurler-Scheie综合征则介于二者之间。
一般出生时表现正常。6个月~1岁后患儿逐渐出现生长缓慢,表情淡漠,反应迟钝,智力低下语言幼稚甚至白痴。大头,前额突出呈舟状,眼距增宽,鼻梁塌陷或扁平,鼻孔增大,唇厚并外翻,张口,舌大且常伸于口外,牙齿小且无光泽齿列稀疏不齐角膜混浊常见,严重者可致失明。常发生中耳炎,并导致听力下降甚至耳聋。心瓣膜及腱索受累可引起心脏增大与心功能不全。支气管软骨病变可致呼吸道狭窄容易并发感染。腹部膨隆,肝脾肿大,多有腹股沟疝或脐疝,可有腹泻或便秘。毛发浓密、粗黑。短颈,耸肩,四肢及躯干短小,脊柱后凸,呈弓形驼背。多数关节呈屈曲状强直活动受限,常有膝踝外翻和扁平足等畸形掌、指粗短可出现腕管综合征。Hurler综合征患者常于儿童期死亡,Scheie综合征及Hurler-Scheie综合征可存活至成年。
骨骼畸形2.黏多糖贮积症Ⅱ型较为少见。根据病情的轻重分为A、B2种亚型,其中A型的病情较重。患者全部为男性,多于2~6岁起病。临床表现与Hurler综合征相似,但出现时间较晚,进展较缓慢智力低下与身材矮小不如Hurler综合征严重。病情严重者从幼儿期开始即有色素性视网膜炎和视盘水肿,但无角膜混浊。听力呈进行性损害,最终发展为耳聋。骨骼畸形较轻微。心脏受累较常见,主要表现为心瓣膜病变、冠心病和充血性心力衰竭多数有阻塞性呼吸暂停综合征,肝脾肿大,腹泻或便秘。患者常于15岁前死亡B型患者病情较轻,有的听力和角膜可均正常亦无骨骼畸形。
3.黏多糖贮积症Ⅲ型临床上极为少见虽然本型可有4种不同的酶缺乏但其临床表现非常相似,主要为进行性的智力减退,其中以黏多糖贮积症ⅢA型的临床进展较快。一般4~5岁以前智力正常其后逐渐出现反应迟钝,智力低下,呈进行性加重。严重者2~3岁即可有智力低下。多有毛发增多。其他方面的改变如特殊面容、身材矮小及骨骼畸形等均不严重甚至可以基本正常。通常有听力损害但无角膜混浊。一般不累及心脏。无腹外疝肝脾可有轻度肿大。身材稍矮或基本正常,极少数可表现为身材矮小。可有关节活动受限甚至有关节强直,手及其他关节可有屈曲畸形。
4.黏多糖贮积症Ⅳ型突出的表现为生长迟缓,一般成年后身高不超过160cm。面容及智力正常学步较晚行走时步态蹒跚不稳。短颈、耸肩。出牙时间较晚,牙列不整齐,牙齿缺乏光泽。角膜混浊可早在儿童期开始出现。听力呈进行性损害。常无心脏受累。肝脾轻度肿大无腹外疝骨骼畸形包括鸡胸、驼背、膝外翻、扁平足及关节屈曲挛缩等畸形,并有明显关节松弛,但无关节强直。可发生颈椎半脱位,引起脊髓压迫症状。多数患者可存活20~30岁。
5.黏多糖贮积症Ⅴ型现认为该型即为黏多糖贮积症Ⅰ型的Seheie型,与Hrular综合征不同之处表现为无严重的角膜混浊,且混浊为周边性,患者智力正常,身材正常或稍矮,寿命基本正常,但有多毛关节强直。背柱、头颅X线示仅有轻微改变。
6.黏多糖贮积症Ⅵ型极为罕见。临床表现与黏多糖贮积症Ⅰ型相似,但患者的智力正常。一般从2~3岁开始出现生长迟缓。颅骨缝闭合较早,可出现脑积水,并引起颅高压症状和痉挛性偏瘫。角膜混浊出现较早,有进行性听力损害严重者有失明和耳聋心脏瓣膜病变肝脾肿大及腹股沟疝等均较为常见。骨骼畸形亦类似于MPSⅠ型但相对较轻,通常上肢长骨受累较下肢严重。关节活动明显受限。可有轻度关节强直。多数患者寿命不超过10岁。
7.黏多糖贮积症Ⅶ型极罕见。特殊面容在出生后不久即开始逐渐出现。一般智力正常角膜混浊及听力损害较常见。多有肝脾肿大,通常不累及心脏,无腹外疝。上肢较短,骨骼发育不良可有鸡胸、膝外翻等骨骼畸形。
粘多糖贮积症_黏多糖贮积症 -科学诊断
根据病史临床表现、实验室检查和X线、CT磁共振、B超、产前检查等手段可确诊。
鉴别诊断:
表2有时各型之间需要进行鉴别诊断主要依据患儿的临床特征与有关的酶学检查详见表2此外黏多糖增多症尚需与以下疾病进行鉴别:
1.多发性硫酸脂酶缺陷症本病的临床表现与黏多糖增多症有相似之处,但智力低下和神经系统症状较黏多糖增多症出现更快常类似于异染性白质萎缩症。患者常有肝肿大和固定的皮肤鱼鳞癣实验室检查无黏多糖尿及细胞酶缺乏。
2.全身性神经节脂苷沉积症(GML神经节脂苷病)兼有脂肪和黏多糖贮积病的临床特点。患儿在婴儿期即有严重的全身神经节脂苷沉积,智能发育迟缓,肌张力低下,肝脾肿大,半数以上的患者有皮肤黄斑和樱红点。
3.甘露糖苷增多症有精神运动发育迟缓,听觉丧失丑陋面容肝脾肿大,肌张力低下,轻度的多发性骨发育不良等。尿中有大量的甘露糖低聚糖,无黏多糖尿。
4.岩藻糖病患者面容丑陋,肝脾肿大,严重的精神、运动发育迟缓,多发性骨发育不良。尿中排泄含有低聚糖的岩藻糖,无黏多糖尿。
5.天门冬酰氨葡萄糖尿症容易与Hurler综合征及Hunter综合征相混淆。患儿出生时正常,逐渐出现宽鼻、塌鼻梁、鼻孔前屈、厚唇等丑陋面容,并有短颈,头颅不对称,脊柱侧凸,肝脾肿大,尿中含有大量的天门冬酰氨葡萄糖。
6.黏脂病黏脂病Ⅰ型的临床表现和X线改变与Hurler综合征有许多共同之处。但黏脂病多数有肌阵挛性抽搐,肌肉萎缩、舞蹈病样手足徐动,眼球震颤以及皮肤黄斑和樱红点。尿中涎酸结合的低聚糖排泄量增加,黏多糖水平正常。
黏脂病Ⅱ型的精神运动发育迟缓发生较早,且发展较快早期有牙龈增生,胸廓狭小,心瓣膜病多见,无角膜混浊,半岁左右即可见长骨骨膜形成,患儿常早年夭折。尿中无黏多糖增多。
黏脂病Ⅳ型亦可有智力发育迟缓、角膜混浊等但无黏多糖尿。
7.Kneist综合征临床表现与Morquio综合征相似,包括大头,鼻梁塌陷,腭裂,短颈,钟状胸,视网膜剥离,听力损害腹外疝肢体和躯干短小,弓形胫骨脊柱后凸,关节强直等。患儿亦可有硫酸角质素尿,但无N-乙酰半乳糖苷-6-硫酸酯酶或β-半乳糖苷酶缺乏
尿液检查实验室检查:
1.尿液检查
(1)黏多糖测定:
①尿黏多糖定性试验:尿斑处呈紫蓝色环状或点状者为阳性,正常人尿斑无颜色改变。
②24h尿黏多糖测定:正常人每天尿中排出的黏多糖为3~25mg。黏多糖增多症患者尿中的黏多糖常超过100mg/24h。由于各类型黏多糖增多症所缺乏的酶不同,其尿中排出的黏多糖成分及数量均有所差异。MPSⅠ、MPSⅡ及MPSⅦ型尿中的黏多糖为硫酸软骨素和硫酸类肝素,其中以Hurler综合征最为显著MPSⅢ型患者尿中只有硫酸类肝素。MPSⅣ型为硫酸角质素,随年龄增大有逐渐减少的趋势。MPSⅥ型主要为硫酸软骨素。
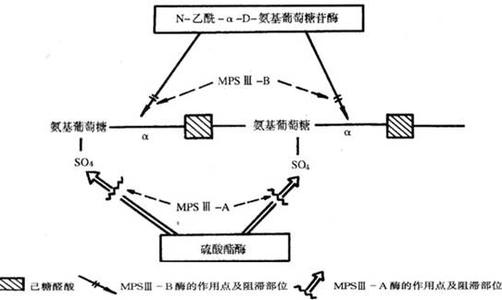
(2)酶活性测定:可测定尿中各种酶的活性,各型黏多糖增多症均有相应的酶活性降低。
2.血液检查
(1)Reilly小体:各型黏多糖增多症均可在末梢血或骨髓的淋巴细胞和中性粒细胞内见有大小不等、形态各异的深紫色黏多糖颗粒,即Reilly小体MPSⅥ型除白细胞以外,尚可在血小板内见到Reilly小体。
(2)酶活性测定:测定末梢血白细胞中的酶活性,是诊断和鉴别各型黏多糖增多症的主要依据。
其它辅助检查:
1.X线检查
(1)MPSⅠ型:在MPSⅠ型的各亚型中,骨骼改变的X线表现亦是以Hurler综合征最为严重。
①头颅:出生后6个月以内基本正常其后逐渐出现颅缝早闭,前囟门闭合延迟。头颅前后径增大呈舟状。脑脊膜增厚可引起阻塞性脑积水可使头颅进一步增大蝶鞍前后径增大,呈仰卧的鞋形;有蛛网膜下囊肿者,可出现蝶鞍增大。颅骨板致密板障增厚,颅底及眶顶亦有硬化。蝶窦、乳突与鼻旁窦发育及气化不良下颌骨粗短,钩状突发育不良,呈扁平或凹陷踝状窝变浅、不规则。牙齿小排列稀疏不齐,磨牙常位于下颌支内。
②脊柱:椎体上下缘呈双凸或椭圆形齿状突短小,可有寰枢关节半脱位。胸椎下段和腰椎上段(胸12、腰1或腰1、腰2)椎体短小呈卵圆形,其前下缘变尖,呈“鸟嘴”样突起,并向后移位形成后凸畸形。
③胸廓:肋骨脊柱端细小,中段至胸骨端逐渐增宽,呈“船桨”样改变。锁骨内侧段明显增粗,外侧段较细并上翘。肩胛骨位置升高,略呈等边三角形,下角变尖,肩胛盂浅而小,甚至消失。肱骨头扁小,颈-干角变小,甚至可呈直角,可有内翻畸形。
④骨盆:髂骨翼外展,髂骨基底部内下方变窄,坐骨闭孔呈椭圆形,耻骨联合增宽。髋臼外上缘呈斜坡状,髋臼变浅,髋臼角增大。股骨头扁小致密,股骨头骺核扁小或不规则且出现时间较晚,股骨颈细长,颈-干角增大呈外翻。
⑤长管骨:上肢改变较下肢明显。由于骨干的塑形障碍,致使骨干粗而短,两端逐渐变细,骨皮质变薄,骨髓腔增大。干骺端可见横条形发育障碍线,骨骺小、不规则,或出现迟延。
⑥短管骨及腕部:掌(跖)、指(趾)近端增粗,远端变尖,呈弹头样。末节指骨(尤其是拇指)远端变尖细,呈爪样屈曲畸形。腕骨不规则,骨化延迟,骨化中心小,且数目少于同龄儿童尺桡骨远侧端发育障碍,腕端关节面呈“Ⅴ”形改变。
(2)MPSⅡ型:骨骼系统改变类似于Hurler综合征,但出现时间相对较晚,进展较慢,改变常较轻。主要改变包括:长骨骨干增宽,多发性骨发育障碍。蝶鞍呈“船桨”样肋骨改变,腰椎呈“鸟嘴”样突出。
(3)MPSⅢ型:本型的骨骼异常较轻微,可有颅顶、颞后部及枕骨增厚乳突气化不良;椎体上下缘稍隆起,或呈椭圆形;锁骨内侧端增宽,部分病人前肋呈“船桨”样增宽;髂骨翼外展,髂骨体短而窄。髋臼上缘较平直;管状骨粗短,干骺端稍增宽,可伴有骨的塑形障碍。骨髓腔窄小、不规则。
(4)MPSⅣ型:头颅、蝶鞍正常。早期椎体略呈圆形其后逐渐变为扁平,前缘正中有舌样突出,椎间隙增宽;齿状突细小或缺如,易引起寰枢关节不稳。胸廓前后径增大,胸骨短缩,并有前突弯曲呈鸡胸状;肋骨前端凹陷,并有增宽、外展,后肋端变细。锁骨内侧端增宽,呈蝶翼状伸向外上方肩胛骨较小,位置升高,肩胛盂变浅或消失。髂骨翼外展,髂骨基底部缩窄,髋臼变浅,由外上向内下呈斜坡状改变,坐骨及耻骨粗短。股骨头干骺端膨大、凹陷、不规则,股骨颈-干角增大,可有髋关节脱位。股骨下端和胫骨上端骨骺扁小,干骺端增宽,呈双重或波浪状致密带,骺线变窄。尺、桡骨远端骨骺小而不规则,甚至消失关节面呈斜坡状;腕骨细小、不规则。长骨普遍粗短,干骺端呈不规则增宽,并有尖角状突起;骨皮质变薄骨小梁稀疏且不规则,骨髓可有缺血性坏死样改变。掌指骨粗短,非骺端变窄。
(5)MPSⅥ型:类似于Hurler综合征。部分患者可有骨骺缺血性坏死样改变以股骨头骨骺多见。
(6)MPSⅦ型:主要为多发性骨发育不良,X线表现与Hurler综合征相似。
2.CT与磁共振(MRI)可准确地了解包括大脑、脊柱骨(软骨)、关节、呼吸道及心血管系统等结构改变的程度和范围。二者均可清楚地显示颅骨发育不良、大脑白质改变、脑积水蛛网膜下腔狭窄、蛛网膜囊肿、颅颈关节的硬脑膜增厚、脊髓压缩等。但在脑白质检查方面,磁共振较CT更为敏感和可靠通常,病程越长则CT与磁共振检查的改变越明显。
3.B型超声用于宫内检查时,可发现胎儿有无骨关节畸形、肝脾肿大和脑积水等异常。
4.组织活检活体组织检查显示肝细胞、皮肤或结缔组织中的成纤维细胞所含的黏多糖代谢酶活性均显著降低。
5.产前检查通常不作为正常妊娠的常规检查。对于生有甘露糖苷增多症患儿的女性,再次怀孕时可行羊水黏多糖浓度及羊水细胞的酶活性测定如果羊水黏多糖浓度明显增高羊水细胞酶活性显著降低,则产前诊断可以确定。
粘多糖贮积症_黏多糖贮积症 -治疗预防
基因治疗本症缺乏彻底根治的方法。虽然在黏多糖增多症的治疗领域已取得了某些进展但大多处于研究阶段,尚未在临床治疗中广泛采用。最有希望治疗黏多糖增多症的方法是特异性的酶替代治疗及基因治疗,二者可改善患者的临床表现以及生存情况。
特异性酶替代治疗可有两种不同的形式。一种是直接给体内输入经过微包裹的酶,此为直接法。另一种则为间接法,即利用反转录病毒进行转基因处理,使患者自体的周围血淋巴细胞或骨髓造血祖细胞逆向转化为含有正常酶基因的细胞或通过骨髓移植给患者体内植入含有正常酶基因的骨髓细胞,从而使患者体内可以自身合成所缺乏的黏多糖代谢酶,上述2种类型的治疗方法均处于临床研究阶段。外科手术主要用于治疗某些躯体和器官的缺陷如心脏瓣膜的置换、角膜移植严重的脊髓压缩等。
预后:
1.黏多糖贮积症Ⅰ型患者病变呈进行性发展,常于15岁左右因心脏病变及呼吸道反复感染而死亡因环枢半脱位,而压迫背髓或背膜,引起颅压升高,甚至危及生命。存活患者均有骨关节功能障碍,并可早发关节退变。
2.黏多糖贮积症Ⅱ型患者存活较长,一般可存活20~30年,病变为进行性发展,病人因心脏、呼吸系统严重受累死亡。
3.黏多糖贮积症Ⅲ型本病有进行性智力减退,骨骼改变常在儿童期可修复,甚至在骨骺融合后可完全消失。但骨关节畸形和塑形异常,则无法完全恢复。少数病人因呼吸道受累而死亡。
4.黏多糖贮积症Ⅳ型很少于儿童期死亡,多数病存活数十年但本病易累及主动脉瓣和呼吸道,致心肺功能不全死亡。患者还可因寰枢关节半脱位引起中枢神经系统严重受损。
5.黏多糖贮积症Ⅶ型重型者常因心脏病变夭折。因关节挛缩可影响到患者正常生活。存活者,均可过早发生继发症、退行性骨关节病。
黏多糖增多症患者的寿命明显缩短,平均预期寿命一般为10~20岁。主要的死亡原因为冠状动脉和主动脉瓣损害以及肺部感染如能早期诊断,并有效减少体内黏多糖的堆积,将有助于患者智力状况的改善,混浊的角膜可恢复透明,肿大的肝脏和脾脏得以缩小阻止骨骼畸形的进一步发展从而大大地改善患者的生活质量,延长寿命。
预防:
黏多糖增多症是一组先天性黏多糖代谢障碍性疾病,属于溶酶体疾病。尚缺乏有关预防资料。
罕见病百科行业百科
概述病例资讯科学研究治疗相关孤儿药信息会议培训医师、专家行业动态药物药品中国地区外国药物政策法规立法国家救助疾病词条罕见病词条分类热搜罕见病词条常见罕见疾病未分类或不明原因中外对比交流社区公益组织交流人群得了罕见病,究竟该去哪里治?疑难病、罕见病不能轻易断定,医生需要反复思考、检查,留意观察病情,对于实在不能解决的疾病才能称为疑难病或罕见病。所以,他说,医生在碰到少见或者有点棘手的疾病时,不要轻易下判断,要从常见病或者多发病着手处理。
瓷娃娃(成骨不全症)疾病简介患有成骨不全症(OI)的儿童以及他们的家庭所面临的问题是复杂的,涉及到解剖、医疗、对残疾的适应和社会等多个层面。其中某些问题是难以克服的,可能无法彻底解决……
哪天是国际罕见病日?由欧洲罕见病组织(EURORDIS)于2008年发起,确定2月29日为国际罕见病日,以这个四年一次的日子意寓罕见病之“罕见”。
什么是罕见病?罕见病,是指盛行率低、少见的疾病,在美国罕见疾病组织所公布的罕见疾病高达一千种之多。
为什么Mayo Clinic这么牛?梅奥医院(英语:Mayo Clinic),是世界著名的医疗机构,位于美国明尼苏达州罗彻斯特(Rochester)。它还有医院设在佛罗里达州的杰克逊维尔(Jacksonville)及亚利桑那州的斯科茨代尔(Scottsdale)。在明尼苏达州、艾奥瓦州、威斯康辛州还有一些小的诊所和医院。
查看“罕见病百科”更多内容>>

