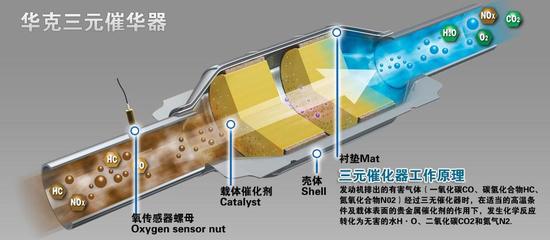
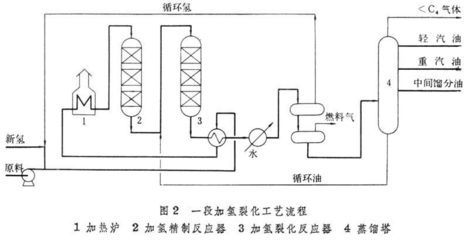
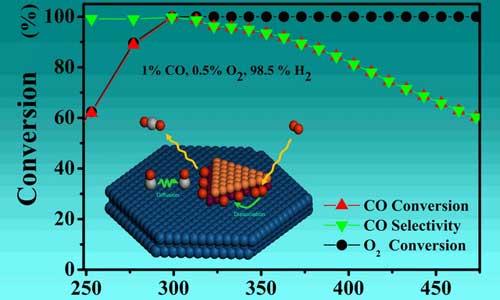
改变化学反应速率而不影响化学平衡的作用。催化剂改变化学反应速率的作用称催化作用,它本质上是一种化学作用。在催化剂参与下进行的化学反应称催化反应。催化是自然界中普遍存在的重要现象,催化作用几乎遍及化学反应的整个领域。
催化_催化 -概述
催化实验
催化即通过催化剂改变反应物的活化能,改变反应物的化学反应速率,反应前后催化剂的量和质均不发生改变的反应。
化学反应物要想发生化学反应,必须使其化学键发生改变,改变或者断裂化学键需要一定的能量支持,能使化学键发生改变所需要的最低能量阈值称之为活化能,而催化剂通过降低化学反应物的活化能而使化学反应更易进行,且大大提高反应速率。
我们知道,在化学反应中能改变化学反应速度而不影响化学平衡的作用称为催化。其中能够改变化学反应速度而本身的组成和数量在反应前后均不发生改变的物质称为催化剂。催化剂可以使化学反应速率增加或减慢,其中增加化学反应速率的催化剂为正催化剂,反之,则为负催化剂。催化作用几乎涉及到化学反应的各个领域。
催化是利用催化剂改变化学反应速度的一种工艺。许多化学工业要利用催化作用来获得需要的反应速度。催化也是一种化工单元过程,催化剂本身在反应中不会被消耗,但催化剂会改变反应速度,一催化剂亦可能参与复数的催化反应。正催化剂可加速反应;负催化剂或抑制剂则会与反应物反应进而降低化学反应。可提高催化剂活性的物质称为促进剂;降低催化剂活性者则称为催化毒。
相较于未催化的反应,同温度的催化反应拥有较低的活化能。催化剂可以借由结合反应物达到极化的效果,如酸催化剂之于羰基化合物的合成;催化剂也可产生非自然的反应中间物,如以四氧化锇催化烯烃的双羟基化中产生的锇酸盐酯;催化剂亦可造成反应物的裂解,如制氢时产生的单原子氢。
很多物质都可以做催化剂,在无机物反应中,通常利用酸、碱、金属或金属化合物作为催化剂,在有机物反应中多用有性的蛋白质分子――酶作为催化剂,生物体内许多化学反应都依赖酶来进行的。
催化_催化 -概念的诞生
催化概念的诞生
古代时,人们就已利用酶酿酒、制醋;中世纪时,炼金术士用硝石作催化剂以硫磺为原料制造硫酸;13世纪,人们发现用硫酸作催化剂能使乙醇变成乙醚。直到19世纪,产业革命有力地推动了科学技术的发展,人们陆续发现了大量的催化现象。
如上所述,催化剂作用是在生产发展的同时为人们由浅入深地认识到的。1835年,贝采里乌斯(Berzelius)首先总结了此前的30多年间发现的催化作用。
为了解释这一现象,他首先采用了“催化”这一名词,并提出催化剂是一种具有“催化力”的外加物质,在这种作用力影响下的反应叫催化反应,即物质的能力“以物质本身存在,而非起亲和力,去唤醒在某一特定温度下还在沉睡的亲和力”。这是最早关于催化反应的理论。
催化_催化 -其认识的深入
认识的深入
人们对于催化作用特点的认识过程是漫长的。在这一认识过程中,许多科学家都亲自从事化学实验并发现了许多催化反应。通过长期实践,逐渐积累加深了认识。
1781年,帕明梯尔(Parmentler)用酸作催化剂,使淀粉水解。1812年,基尔霍夫(Kirchhoff)发现,如果有酸类存在,蔗糖的水解作用会进行得很快,反之则很缓慢。而在整个水解过程中,酸类并无什么变化,它好像并不参加反应,只是加速了反应过程。同时,基尔霍夫还观测到,淀粉在稀硫酸溶液中可以变化为葡萄糖。1817年,戴维(Davy)在实验中发现铂能促使醇蒸气在空气中氧化。1838年,德拉托和施万分别都发现糖之所以能发酵成为酒精和二氧化碳,是由于一种微生物的存在。贝采里乌斯就此提出,在生物体中存在的那些由普通物质、植物汁液或者血而生成无数种化合物,可能都是由此种类似的有机体组成。后来,居内将这些有机催化剂称为“酶”。
1850年,威廉米(Wilhelmy)通过研究酸在蔗糖水解中的作用规律,第一次成功地分析了化学反应速度的问题,从此开始了对化学动力学的定量研究。1862年,圣・吉尔和贝特罗在实验中发现,如果按照分子比将醋酸乙脂与水混合,经过几星期之后于进行观测,发现醋酸乙脂已部分水解成为乙醇和醋酸。这一反应的速度随时间处长而呈递减趋势。再将乙醇与酸混合,反应生成了醋酸乙脂,平衡后的比例相同。这一反应的速度同样很慢。但是,当有无机酸存在时,上述两个反应则可在几小时内完成。这样,无机酸作为一种催化剂可以促进两个反应向任一方向进行的反应速度。
1884年前后,包括奥斯特瓦尔德(Ostward)在内的几位化学家研究了各种酸对酯的水解作用以及蔗糖转化等现象的酸碱催化作用的解释,他认为催化剂现象的本质,在于某些物质具有一种特别强烈的使原本没有它参加而速度很慢的反应加速的特殊性能。
1895年奥斯特瓦尔德提出的催化定义为:“催化剂是一种物质,其能改变某一反应的反应速率而不能改变该反应的能量因素。他说,任何物质,如果它不参加到化学反应的最终产物中去,只是改变这个反应的速度即称为催化剂。另外,他通过总结大量的实验结果,根据热力学第二定律,提出了平衡的达成,不能改变平衡常数。
1905年,勒・罗西诺(LeRosignol)和哈伯(Haber)等人,根据化学热力学的原理,研究计算了氢、氮和氨在各种温度和压力平衡情况后,利用各种催化剂的帮助,研究出从空气中的氮合成氨的实验方法。催化与可逆过程的关系也有研究,催化不影响平衡状态概念得以确立。从催化与平衡的讨论中可以得出结论:在任何可逆反应中催化剂可加速正反应,也可加速逆反应,比如加氢催化剂在逆反应脱氢中也是有效的。
催化_催化 -其理论的发展
理论的发展
在寻找催化剂和催化反应的过程的同时积累了大量的资料,使人们对催化剂和催化作用的认识不断深入。关于催化反应的理论也逐步得以发展。催化剂为什么能够改变化学反应的速度,而它本身在反应后又不发生化学变化呢?为了解释这一问题,在19世纪初期,就已经有人提出关于催化剂在反应中生成中间化合物的假说,认为催化剂之所以有所谓“催化能力”,是由于生成了中间化合物的结果。
1806年,德索尔姆(Desormes)和克雷蒙(Clement)研究一氧化氮对二氧化硫氧化的催化作用时,推想一氧化碳先与大气中的氧反应生成某种中间化合物。这一中间化合物再与二氧化硫相互作用,此时把氧转交给后者,中间物质自身又变为一氧化氮。一氧化氮可以再被空气氧化,之后再把氧交给二氧化硫。如果按照这种概念,这种均相催化反应是交错地进行的氧化还原过程的综合。一个缺憾是他们没有提出具体的反应的具体过程。
1806年,德索尔姆(Desormes)和克雷蒙(Clement)研究一氧化氮对二氧化硫氧化的催化作用时,推想一氧化碳先与大气中的氧反应生成某种中间化合物。这一中间化合物再与二氧化硫相互作用,此时把氧转交给后者,中间物质自身又变为一氧化氮。一氧化氮可以再被空气氧化,之后再把氧交给二氧化硫。如果按照这种概念,这种均相催化反应是交错地进行的氧化还原过程的综合。一个缺憾是他们没有提出具体的反应的具体过程。
1835年,贝采里乌斯提出的过程与克雷蒙和德索尔姆的概念最为类似,他认为催化反应由下列两个过程交替进行:
2NO+O=N2O3
SO2+N2O3+H2O=H2SO4+2NO
可以看出,在贝采里乌斯所提出的过程中,三氧化氮就是相当于克雷蒙和德索尔姆所推想的把空气中的氧转交给二氧化硫的活性中间物质。在贝采里乌斯之后,威廉逊(Willianson)曾于1851年用相似的方法来解释该反应的进行。从此,中间化合物这一概念得到确立,并在以后得到广泛应用。
邢歇伍德(Hinshelwood)等人在1930年,以碘蒸气为催化剂进行乙醛蒸气的加热分解反应,发现均相催化反应的速度常常与催化剂的深度成正比的。而在该反应中,作为催化剂的碘蒸气的深度始终不变,邢歇伍德认为,这一事实说明由于催化剂K先与某一反应物A或B相互作用,生成了活性的中间化合物X,此中间化合物进一步转变而生成C并使催化剂再生。他们用以下形式表达上述反应历程:
A+K=X+……
X+B=C+K……
可见,活性的中间化合物的假说因此得以进一步的证实和完善,同时均相催化理论也得到了发展。
随着更多实验事实的发现和研究的不断伸入,人们发现催化剂作用不仅是均相地进行,更多的是这一类反应则是在多相中进行。并且,这时反应物在相界面上的浓度更大,这种现象被称为“吸附作用”。科学家们把吸附分为两种类型,一种是简单的物理吸附;另一种是吸附的同时形成化学键,称为化学吸附,当然,这一类完成是吸附的同时形成化学键,称为化学吸附,当然,这一类完成的过程是曲折的。
催化反应的吸附理论首先是由意大利人珀兰尼(Polanyi)在1824年提出的。他认为,由于吸附作用使物质的质点相互接近,因而它们之间容易发生反应。他说,吸附作用是由于电力而产生的分子吸引力。
理论的发展
1834年,法拉第(Faraday)则提出了与上者不同的吸附理论,他认为催化反应不是电力使然,而是靠体物质相互吸收所产生的气体张力。他认为,如果催化剂表面极为干净,气体就会附着其上而凝结,一部分反应分子彼此接近到一定程度时,就会使新合力发生作用,抵消排斥力,因而使反应变得容易进行。朗缪尔(Langmuir)在1916年间,发表了一系列关于单分子表面膜的行为及性质,和关于固体表面吸附作用的研究成果影响到催化理论的形成。之后,科学界在1920年-1940年间大量的研究成果对催化吸附理论有着重大影响。
值得注意的是,在这一时期通过对吸附量和脱附速度的研究,以及关于催化过程中催化失去催化活力的研究,得出了对多相催化理论有着根本意义的结论,即催化反应是在催化剂表面直接相连的单分子层中进行的。
就此,美国人泰勒(Taylor)于1925年首先提出了活性中心理论,其出发点即催化剂失去活性这一实验事实。他认为催化剂的表面是不均匀的,位于催化剂表面微型晶体的棱和顶角处的原子具有不饱合的键,因而形成了活性中心,催化反应只发生在这一活性中心。泰勒的理论很好地解释了催化剂制备对活性的影响以及毒物对活性的作用。
在泰勒之后,前苏联的两位科学家对活性中心理论进行了进一步的完善和发展。1929年,巴兰金(Баландин)提出了多位催化理论,认为催化剂活性中心的结构应当与反应物分子在催化反应过程中发生变化的那部分结构处于向何对应。这一理论把催化活化看作反应物中的多位体的反应过程,并且这个作用会引起反应物中价键的变形,并使反应物分子活化,促成新价键的形成。另一位苏联人柯巴捷夫(н.И.Koбазeв)于1939年提出了活性集团理论,与泰勒不同的是他认为活性中心是催化剂表面是上非晶体中几个催化剂原子组成的集团。
20世纪50年代以后,随着固体物理的发展,催化的电子理论应运而生。在这一层面上,科学家们得到了丰富的实验成果,他们将金属催化性质与基电子行为和甲子能级联系起来。70年代,根据催化剂表面的原子结构、络合物中金属原子簇的结构和性质,利用量子化学理论,对多相催化的高分散的金属催化剂活性集团产生催化活性的根源进行研究。在科学突飞猛进的今天,催化作用的实质以及催化剂发生作用的秘密正逐渐为人们所认知。
二次世界大战前化学工业基于煤炭工业,着重于合成氨、硝酸生产、油脂硬化及甲醇合成,20世纪50年代初期,石油化学工业的发展迅速,导致许多新燃料的面市、聚合物工业的突起、选择氧化、氢甲酰化和其它均相反应也逐渐变得重要。20世纪70年代石油危机使人们更加珍惜能源和原材料的合理利用。汽车尾气和工业排放污染治理受到重视。催化在这些方面起着决定作用。
约有80%的化学过程使用了催化剂,它的销售额接近100亿美元,但却只有其所创造产品收入的1%的催化剂市场正以每年约10%的速度递增。
催化_催化 -相关种类
催化示意图
均相催化
催化剂与反应物同处于一均匀物相中的催化作用。有液相和气相均相催化。液态酸碱催化剂,可溶性过渡金属化合物催化剂和碘、一氧化氮等气态分子催化剂的催化属于这一类。均相催化剂的活性中心比较均一,选择性较高,副反应较少,易于用光谱、波谱、同位素示踪等方法来研究催化剂的作用,反应动力学一般不复杂。但均相催化剂有难以分离、回收和再生的缺点。
多相催化
多相催化发生在两相的界面上,通常催化剂为多孔固体,反应物为液体或气体。多相催化反应通常可按下述七步进行:①反应物的外扩散──反应物向催化剂外表面扩散;②反应物的内扩散──在催化剂外表面的反应物向催化剂孔内扩散;③反应物的化学吸附;④表面化学反应;⑤产物脱附;⑥产物内扩散;⑦产物外扩散。这一系列步骤中反应最慢的一步称为速率控制步骤。化学吸附是最重要的步骤,化学吸附使反应物分子得到活化,降低了化学反应的活化能。因此,若要催化反应进行,必须至少有一种反应物分子在催化剂表面上发生化学吸附。固体催化剂表面是不均匀的,表面上只有一部分点对反应物分子起活化作用,这些点被称为活性中心。
生物催化
酶是一种生物催化剂,生物体内的所有化学变化几乎都是在酶催化下进行的,酶的催化作用称为生物催化。酶的催化活性高,选择性强。生物催化在常温中性条件下进行,高温、强酸和强碱都会使酶丧失活性。离体的酶仍具有催化活性,可制成各种酶制剂应用在医学和工农业生产上。其他还有电催化、光助催化、光电催化等。按催化作用机理,可把催化剂分为金属催化剂、金属氧化物催化剂、配位(络合)催化剂、酸碱催化剂和多功能催化剂。
金属催化
金属催化剂主要用于脱氢和加氢反应。有些金属还具有氧化和重整的催化活性。金属催化剂主要是指4、5、6周期的某些过渡金属,如铁、金、铂、钯、铑、铱等。金属催化主要决定于金属原子的电子结构,特别是没有参与金属键的d轨道电子和d空轨道与被吸附分子形成吸附键的能力。因此,金属催化剂的化学吸附能力和d轨道百分数是决定催化活性的主要因素。
金属氧化物催化
主要是指过渡金属氧化物催化,非过渡金属氧化物催化已归入酸碱催化。过渡金属氧化物催化剂广泛用于氧化、加氢、脱氢、聚合、加合等反应。实用的金属氧化物催化剂常为多组分氧化物的混合物,很多金属氧化物催化剂是半导体,其化学组成大多是非化学计量的,因此,催化剂组分很复杂。金属氧化物催化剂的导电性和逸出功、金属离子的d电子组态、氧化物中晶格氧特性、半导体电子能带、催化剂表面吸附能力等,都与催化剂的催化活性有关。
配位(络合)催化
金属、特别是过渡金属及其化合物有很强的络合能力,能形成多种类型的络合物。某些分子与金属(或金属离子)络合后便易于进行某特定反应,该反应称为配位(络合)催化反应,该金属或其化合物起络合催化剂作用。过渡金属络合催化剂在溶液中作为均相催化剂方面的研究和应用较多。过渡金属络合催化作用一般都是配位(络合)催化作用,即催化剂在其空配位上络合活化反应物分子。络合催化剂一般都是金属络合物或化合物,如钯、铑、钛、钴的络合物等。
酸碱催化
阿伦尼乌斯酸碱、布仑斯惕酸碱、路易斯酸碱(见酸碱理论)的催化作用都属酸碱催化作用。酸碱催化可分均相催化和多相催化。许多离子型有机反应,如水解、水合、脱水、缩合、酯化、重排等,常可用酸碱均相催化。固体酸催化剂广泛用于催化裂化、异构化、烷基化、脱水、氢转移、歧化、聚合等反应。
多功能催化
若反应物A直接变成产物B的反应难以进行,则可通过几个催化化学反应来实现。例如SⅠ、SⅡ分别为两个反应的催化剂,则可将SⅠ、SⅡ混合起来制成双功能催化剂,使A→B的反应实现。这就是多功能催化。多功能催化与一个催化反应的多步骤是有区别的,此处的C不是在催化剂表面形成的中间络合物,而是由SⅠ表面脱附出来的、有其自己的结构和热力学性质的化学物质。应用现代化学工业的巨大成就与催化剂的使用是分不开的。约80%以上的化学工业产品是借助于催化过程来生产的,可以说,没有催化剂就没有现代化学工业。
催化_催化 -其原理
催化原理
降低活化能
在催化反应过程中,至少必须有一种反应物分子与催化剂发生了某种形式的化学作用。由于催化剂的介入,化学反应改变了进行途径,而新的反应途径需要的活化能较低,这就是催化得以提高化学反应速率的原因。例如,化学反应A+B─→AB,所需活化能为E,在催化剂C参与下,反应按以下两步进行:
A+C─→AC,所需活化能为E1
AC+B─→AB+C,所需活化能为E2
E1、E2都小于E(见图)。催化剂C只是暂时介入了化学反应,反应结束后,催化剂C即行再生。
按阿伦尼乌斯方程k=Ae-E/RT(式中A为指前因子;R为气体常数;T为热力学温度),以反应速率常数k表示的反应速率主要决定于反应活化能E,若催化使反应活化能降低ΔE,则反应速率即提高e-ΔE/RT倍。催化反应一般能降低活化能约10千卡/摩尔,若反应在300K下进行,则反应速率可增加约1.7×10倍。
催化剂表面性能
多相催化是工业上应用最多的,这种催化作用在催化剂表面上进行,因此,固体催化剂的表面性质对催化作用就会有很大影响。催化剂比表面积大,表面上活化中心点多,表面对反应物吸附能力强,这些都对催化活性有利,因为化学吸附能降低反应活化能。表面孔隙度大和孔径大小合适对催化剂的选择性有利,例如分子筛催化剂的极强的选择性,就是由于它的孔径尺寸只能允许某种分子进入孔内,到达催化剂表面而被催化。
催化剂的作用方式
催化剂与反应物分子怎样发生化学作用,这与催化剂和反应物分子本身的性质有关。实验证明:有机化合物的酸催化反应一般是通过正碳离子机理进行的;碱催化反应则是由OH、RO、RCOO等阴离子起催化作用,例如:
CH3COOC2H5+OH─→CH3COOH+C2H5O
C2H5O+H2O─→C2H5OH+OH
过渡金属化合物催化剂在均相催化反应中的作用是络合催化作用;醇钠催化丁二烯聚合是通过自由基机理进行的,例如:
C4H6+NaR─→R?+C4H6Na?
C4H6Na?+nC4H6─→Na(C4H6)n+1
催化剂的性能指标
①催化活性:催化剂参与了化学反应,降低了化学反应的活化能,大大加快了化学反应的速率。这说明催化剂具有催化活性。催化反应的速率是催化剂活性大小的衡量尺度。活性是评价催化剂好坏的最主要的指标。②选择性:一种催化剂只对某一类反应具有明显的加速作用,对其他反应则加速作用甚小,甚至没有加速作用。这一性能就是催化剂选择性。催化剂的选择性决定了催化作用的定向性。可通过选择不同的催化剂来控制或改变化学反应的方向。③寿命或稳定性:催化剂的稳定性以寿命表示。它包括热稳定性、机械稳定性和抗毒稳定性。
催化_催化 -工艺单元组成
有机废气催化燃烧净化设备
催化燃烧的工艺组成不同的排放场合和不同的废气,有不同的工艺流程。但不论采取哪种工艺流程,都由如下工艺单元组成。
①废气预处理为了避免催化剂床层的堵塞和催化剂中毒,废气在进入床层之前必须进行预处理,以除去废气中的粉尘、液滴及催化剂的毒物。
②预热装置预热装置包括废气预热装置和催化剂燃烧器预热装置。因为催化剂都有一个催化活性温度,对催化燃烧来说称催化剂起燃温度,必须使废气和床层的温度达到起燃温度才能进行催化燃烧,因此,必须设置预热装置。但对于排出的废气本身温度就较高的场合,如漆包线、绝缘材料、烤漆等烘干排气,温度可达300℃以上,则不必设置预热装置。预热装置加热后的热气可采用换热器和床层内布管的方式。预热器的热源可采用烟道气或电加热,目前采用电加热较多。当催化反应开始后,可尽量以回收的反应热来预热废气。在反应热较大的场合,还应设置废热回收装置,以节约能源。预热废气的热源温度一般都超过催化剂的活性温度。为保护催化剂,加热装置应与催化燃烧装置保持一定距离,这样还能使废气温度分布均匀。从需要预热这一点出发,催化燃烧法最适用于连续排气的净化,若间歇排气,不仅每次预热需要耗能,反应热也无法回收利用,会造成很大的能源浪费,在设计和选择时应注意这一点。
③催化燃烧装置一般采用固定床催化反应器。反应器的设计按规范进行,应便于操作,维修方便,便于装卸催化剂。在进行催化燃烧的工艺设计时,应根据具体情况,对于处理气量较大的场合,设计成分建式流程,即预热器、反应器独立装设,其间用管道连接。对于处理气量小的场合,可采用催化焚烧炉见图16-13,把预热与反应组合在一起,但要注意预热段与反应段间的距离。
在有机物废气的催化燃烧中,所要处理的有机物废气在高温下与空气混合易引起爆炸,安全问题十分重要。因而,一方面必须控制有机物与空气的混合比,使之在爆炸下限;另一方面,催化燃烧系统应设监测报警装置和有防爆措施。
催化_催化 -所需原料
催化剂
助催化剂:本身不具有催化活性,但加入后(加入量一般低于催化剂量的10%)可显著提高催化剂的活性、选择性和稳定性。一种工业催化剂往往要加入几种助催化剂,才能使催化剂的活性、选择性和寿命都达到预定要求。例如,合成氨用的双促进催化剂铁-三氧化二铝-氧化钾中的三氧化二铝和氧化钾就是助催化剂。
抑制剂:催化剂中含有的能降低催化剂活性的物质。在多数场合,它是在催化剂制备过程中由原料不可避免地带入的。在催化反应过程中,由反应物带入的抑制剂则称催化毒物。
催化剂载体:不具有催化活性的用来负载催化剂的固体物质。把催化剂成分分散负载在载体上制成的催化剂称负载型催化剂。常用的催化剂载体有活性碳、硅藻土、活性氧化铝、硅胶和分子筛。对载体的要求是机械强度高、热稳定性和化学稳定性好。载体虽不具有催化活性,但它可能与催化剂发生化学作用,载体也改变了催化剂的表面性能,因而选用合适的载体,也可以提高催化剂的活性、选择性和寿命。
由于有机物催化燃烧的催化剂分为贵金属以铂、钯为主和贱金属催化剂。贵金属为活性组分的催化剂分为全金属催化剂和以氧化铝为载体的催化剂。全金属催化剂是以镍或镍铬合金为载体,将载体做成带、片、丸、丝等形状,采用化学镀或电镀的方法,将铂、钯等贵金属沉积其上,然后做成便于装卸的催化剂构件。
由氧化铝作载体的贵金属催化剂,一般是以陶瓷结构作为支架,在陶瓷结构上涂覆一层仅有0.13mm的α-氧化铝薄层,而活性组分铂、钯就以微晶状态沉积或分散在多孔的氧化铝薄层中。
但由于贵金属催化剂价格昂贵,资源少,多年来人们特别注重新型的、价格较为便宜的催化剂的开发研究,我国是世界上稀土资源最多的国家,我国的科技工作者研究开发了不少稀土催化剂,有些性能也较好。
催化_催化 -相关提示
催化剂
在催化剂使用过程中,由于体系中存在少量的杂质,可使催化剂的活性和选择性减小或者消失,这种现象叫催化剂中毒。这些能使催化剂中毒的物质称之为催化剂毒物,这些毒物在反应过程中或强吸附在活性中心上,或与活性中心起化学作用而变为别的物质,使活性中心失活。毒物通常是反应原料中带来的杂质,或者是催化剂本身的某些杂质,另外,反应产物或副产物本身也可能对催化剂毒化,一般所指的是硫化物如H2S、硫氧化碳、RSH等及含氧化合物如H2O、CO2、O2以及含磷、砷、卤素化合物、重金属化合物等。
毒物不单单是对催化剂来说的,而且还针对这个催化剂所催化的反应,也就是说,对某一催化剂,只有联系到它所催化的反应时,才能清楚什么物质是毒物。即使同一种催化剂,一种物质可能毒化某一反应而不影响另一反应。按毒物与催化剂表面作用的程度可分为暂时性中毒和永久性中毒。暂时性中毒亦称可逆中毒,催化剂表面所吸附的毒物可用解吸的办法驱逐,使催化剂恢复活性,然而这种可再生性一般也不能使催化剂恢复到中毒前的水平。永久性中毒称不可逆中毒,这时,毒物与催化剂活性中心生成了结合力很强的物质,不能用一般方法将它去除或根本无法去除。
催化剂的老化主要是由于热稳定性与机械稳定性决定的,例如低熔点活性组分的流失或升华,会大大降低催化剂的活性。催化剂的工作温度对催化剂的老化影响很大,温度选择和控制不好,会使催化剂半熔或烧结,从而导致催化剂表面积的下降而降低活性。另外,内部杂质向表面的迁移,冷热应力交替所造成的机械性粉末被气流带走。所有这些,都会加速催化剂的老化,而其中最主要的是温度的影响,工作温度越高,老化速度越快。因此,在催化剂的活性温度范围内选择合适的反应温度将有助于延长催化剂的寿命。但是,过低的反应温度也是不可取的,会降低反应速率。
为了提高催化剂的热稳定性,常常选择合适的耐高温的载体来提高活性组分的分散度,可防止其颗粒变大而烧结,例如以纯铜作催化剂时,在200℃即失去活性,但如果采用共沉积法将Cu载于Cr2O3载体上,就能在较高的温度下保持其活性。