Wilson氏病是一种较少见的常染色体隐性遗传的铜代谢疾病。也称:肝豆状核变性(hepatolenticular degeneration,HLD)又称威尔逊氏病。
wilson病_WILSON 氏病 -概述
wilson's病wilson's disease:切面可见较大结节,左叶有坏死灶威尔逊氏病(Wilson’s Disease)是一种较少见的常染色体隐性遗传的铜代谢疾病。
早自Frerichs(1861)、Westphal(1883)和Strumpel(1898)先后发现一组病例,临床酷似多发性硬化的表现,而尸检却缺乏中枢神经系统特征性的硬化斑,命名为假性硬化症。
1911年Wilson证实青少年发病的假性硬化症,其病理特征是肝硬化和大脑基底节区的豆状核变性,命名为进行性肝豆状核变性。1921年Hall定名肝豆状核变性(HepatolenticularDegeneration,HLD)或威尔逊氏(Wilson)假性硬化症,后人又称为威尔逊氏病(Wilson’sdisease,WD)。
据欧美流调统计,本病发病率为0.2/10万人口,患病率为1/10万人口,杂合子为1/4000人口。日本资料患病率约1.9~6.8/10万人口,杂合子高达6.6~13/1000人口。国内虽缺乏本病的流调资料,但本中心(安徽中医学院神经病学研究所附属医院)1976年10月至2000年10月间收治来自全国各地的HLD患者已近3000例,可见本病在中国并不少见。
本病已明确属常染色体隐性遗传性铜代谢障碍,造成铜在体内各脏器尤以大脑豆状核、肝脏、肾脏及角膜大量沉着,而由于铜离子在各脏器沉积的先后不同和数量不一,临床出现多种多样的临床表现,如震颤、扭转痉挛、精神障碍、肝脾肿大、腹水等。祖国医学分别归属于“颤症”、“癫狂”、“黄疸”、“积聚”、“鼓胀”等范畴。
wilson病_WILSON 氏病 -病因
威尔逊氏病(Wilsonsdisease,WD),是一种常染色体隐形遗传的铜代谢缺陷病,其基因定位于13q14.3,编码1个P型ATP酶,此酶参与铜跨膜转运的代谢过程。目前研究多认为由于WD基因突变使其功能降低或丧失而导致铜代谢异常,肝合成铜蓝蛋白速度减慢,胆汁排铜明显减少,铜沉积于肝、脑、肾、角膜、血细胞和关节等组织中,引起了相应脏器损害的临床症状。铜蓄积可导致肝细胞坏死、肝纤维化;从坏死的肝细胞释放的大量铜可导致溶血,并逐渐沉积在脑、肾、角膜、骨关节部位,引起多器官受累。不同程度的肝细胞损害,脑退行性病变和角膜边缘有铜盐沉着(K-F环)为其临床特征。早期诊断和治疗可避免严重的不可逆的组织器官损害,常可以获得与健康人一样的生活和寿命。1921年Hall首先分析68例有血缘关系的HLD病人,其中34例至少与另一名患者有血缘关系,且绝大多数是表(堂)亲。因此Hall确信HLD属遗传性疾病,并且指出是由2个不同的隐匿性代谢缺陷的基因传递所致,其传递形式与常染色体隐性遗传相一致。作者等调查167例HLD先证者的家系,615位先证者及其同胞中,278人证实为HLD,按Hardy-Weinberg公式计算;(278-167)/(615-167)=0.248。此值与常染色体隐性遗传的预期值0.25极接近,统计学无显著差异(P>0.05),符合常染色体隐性遗传。
wilson病_WILSON 氏病 -临床表现
wilson's病 wilson's disease:肝切面呈结节样,左叶见坏死灶。一、症状与体征
临床主要表现神经精神症状与肝脏症状两大方面。欧美报道,约70%的WD患者以神经症状为首发症状,肝脏症状次之。但我们统计近7年1011例患者以神经症状起病者480例(47.48%),肝脏症状起病者404例(39.96%),其次为骨关节及肾脏损害症状。
(一)神经精神症状
1.震颤早期常限于上肢,渐延及全身.多表现为快速、节律性,粗大似扑翼样的姿位性震颤,可并有运动时加重的意向性震颤。
2.发音障碍与吞咽困难多见于儿童期发病的HLD.说话缓慢似吟诗,或音调平坦似念经,或言语断辍似呐吃;也可含糊不清、暴发性或震颤性语言.吞咽困难多发生于晚期患者。
3.肌张力改变大多数患者肌张力呈齿轮样、铅管样增高,往往引致动作迟缓、面部表情减少、写字困难、步行障碍等。少数舞蹈型患者伴肌张力减退。
4.癫痫发作较少见。总结418例HLD中,11例(2.6%)于病程中出现不同类型癫痫发作,其中10例为全身强直-阵挛发作或部分性运动发作,仅1例为失神发作。
5.精神症状早期病人智能多无明显变化,但急性起病的儿童较早发生智力减退;大多数HLD具有性格改变,如自制力减退、情绪不稳、易激动等;重症可出现抑郁、狂躁、幻觉、妄想、冲动等,可引起伤人自伤行为。少数患者以精神症状为首发症状,易被误诊为精神分裂症。
(二)肝脏症状以肝脏症状为首发症状有:
1.通常约5~10岁发病。由于肝脏内铜离子沉积达超饱和,引起急性肝功能衰竭,即腹型肝豆状核变性.临床表现为,全身倦怠、嗜睡、食欲不振、恶心呕吐、腹部膨胀及高度黄疸,病情迅速恶化,多于一周至一月左右死亡,往往在其同胞被确诊为HLD后,回顾病史时方考虑本病可能。
2.约半数患者在5~10岁内,出现一过性黄疸、短期谷丙转氨酶增高或/及轻度腹水,不久迅速恢复。数年后当神经症状出现时,肝脏可轻度肿大或不能扪及,肝功能轻度损害或正常范围,但B超检查已有不同程度损害。
3.少儿期缓慢进行食欲不振、轻度黄疸、肝脏肿大和腹水,酷似肝硬化的表现.经数月至数年,消化道症状迁延不愈或日益加重,而渐渐出现震颤、肌僵直等神经症状.神经症状一旦出现,肝症状迅速恶化,多于几周至2~3月内陷入肝昏迷.因此对原因不明的肝硬化少儿患者。
4.部分青少年患者可表现缓慢进行脾脏肿大,并引致贫血、白细胞或(及)血小板减少等脾功能亢进征象,一般在脾切除或/及门脉分流术后不久,出现神经症状并迅速恶化,常于短期内死亡;少数患者因食管静脉破裂致上消化道出血而迅速促发神经症状。
5.肝症状发生于其他症状后
(1)先出现神经症状,长期误诊或不规则驱铜治疗,神经症状迁延达晚期,渐发生黄疸、腹水乃至肝昏迷。
(2)以神经症状获得正确诊断,体检时才发现轻度肝脾肿大或/及肝功能异常。
(三)角膜色素环(Kayser-Fleisher环,K-F环)肉眼或裂隙灯在角膜后弹力层周边部可见棕色、灰色环。
二、临床分型
(一)潜伏型(亚临床型)一般为先证者的一级亲属,在进行铜代谢筛选检查时发现。
(二)显性型(临床表现型)
1.脑型(以神经症状为核心症状)
(1)广义肝豆状核变性型临床特征为:①一般于15岁以前发病;②肌僵直显著,震颤轻;③晚期呈全身扭转痉挛。
(2)舞蹈-手足徐动型儿童多见,以脸面不自主扭动和四肢不规则、快速舞动伴肢端缓慢扭动为特征,少数呈投掷样运动。
(3)假性硬化型:临床特征为:①大多于20岁以后起病;②全身震颤较明显,而肌僵直较轻。
(4)精神障碍型以重精神症状为首发症状,神经症状较轻或缺如,常易误诊为精神分裂症等各种重精神病。
2.脊髓型或脑脊髓型此型极少见,脊髓型临床特征为:①多见于10~20岁男性患者;②对称性痉挛性截瘫为主要表现.如伴有意识不清、言语错乱和震颤等脑症状,称脑脊髓型。
3.骨-肌型临床特征为:①大多于17~18岁左右发病;②明显骨关节症状及四肢近端肌无力、肌萎缩;③神经症状和肝症状较轻或缺如;④病情发展缓慢,预后较良好。但作者长期随访发现,晚期也可出现肌僵直等锥体外系或/及肝脏症征。
4.腹型临床特征为:①5~10岁前常以发热、严重黄疸、中度腹水突然起病;②进展迅速,往往在一周至一月内死亡;③生前无锥体外系症状。
5.脑-内脏混合型本组以肝型最多见,临床表现多样,主要有下列特征:①青少年发生缓慢进行性脾肿大,酷似门脉高压症;②儿童期发生原因不明的进行性肝硬化.如晚期出现神经症状,称脑-肝型。另一部分脑型患者,不恰当或不规则治疗而达晚期,常并有肝硬化等肝症状,亦属脑-肝型范畴。此外,以浮肿、蛋白尿、血尿为主证者,称肾型;以明显脑症状并心脏症状为主证者称脑-心型。
wilson病_WILSON 氏病 -诊断

一、诊断标准
肝豆状核变性诊断标准:
1.家族遗传史:父母是近亲婚配、同胞有HLD患者或死于原因不明的肝病者。
2.缓慢进行性震颤、肌僵直、构语障碍等锥体外系症状、体征或/及肝症状。
3.肉眼或裂隙灯证实有K-F环。
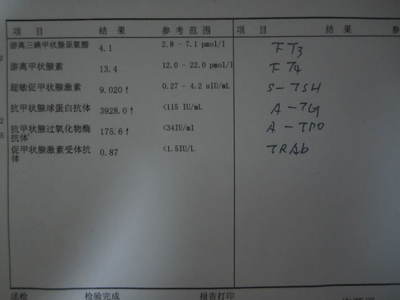
4.血清铜蓝蛋白<200mg/L或铜氧化酶<0.2OD。
5.尿铜>1.6μmol/24h。
6.肝铜>250μg/g(干重)。
判断:①凡完全具备上述1~3项或2及4项者,可确诊为临床显性型。②仅具有上述3~5项或3~4项者属无症状型HLD。③仅有1、2项或1、3项者,应怀疑HLD。
凡具下列情况应高度怀疑HLD患者,都必须行裂隙灯检查有无角膜K-F环和铜代谢测检。
1.已证实HLD患者的同胞。
2.同胞中有幼年死于暴发性肝炎或其他肝病(尤其病毒性肝炎血清抗原抗体阴性)者。
3.儿童或少年发生原因不明的肝硬变、一过性黄疸、流涎、震颤、舞蹈样运动或精神错乱,均需注意与HLD鉴别,必要时,需进一步行裂隙灯和铜代谢检查。
二、鉴别诊断
1.肝型HLD需与慢性活动性肝炎、慢性胆汁郁滞综合征或门脉性肝硬变等肝病鉴别。但肝病无血清铜减低、尿铜增高、血清铜蓝蛋白和铜氧化酶显著降低等铜代谢异常;亦无角膜K-F环。
2.假性硬化型HLD需与帕金森病鉴别,肝豆状核变性型HLD需与特发性肌张力障碍鉴别。但帕金森病、特发性肌张力障碍均无铜代谢异常及角膜K-F环,可与HLD区别。
wilson病_WILSON 氏病 -治疗
本病目前可以治疗,但无法根治。一、低铜饮食
减少食物中铜的摄食为治疗的重要组成部分。
二、中医治疗
在30多年的研究期间,肝豆汤(片)共治疗HLD患者近3000例,观察表明其对为临床症状有明显的缓解作用。临床和实验研究均表明该方具有显著的尿及胆汁排铜作用;在长期的临床实践中我们逐步发现HLD患者除多数存在热象以外,尚有其它中医证型。为此,拟订了肝豆汤(片)II号,临床获得良疗效,为HLD提供了一种有效、低毒、费用低廉的治疗方法。
三、西医治疗
(一)二巯基丙醇(dimercaprolum)
BAL是含双巯基的化合物,水溶液不稳定,故配成10%油剂溶液,仅供肌内注射,已趋向淘汰。。缺点是:①副反应多,如臀部脓肿、肝功能损害等使病人被迫停止治疗;长期连续应用,排铜作用逐渐衰减。
(二)二巯丁二酸(dimercaptosuccinicacid,DMSA)和二巯丁二酸钠(sodiumdimercaptosuccinate,Na-DMS)Na-DMS静注后,血浓度迅速达高峰,4小时排泄80%,无蓄积作用。优点为:①Na-DMS排铜量较高,不仅尿排铜量较疗前平均增高7.7±1.4μmol/24h且胆汁排铜平均增加1.5倍;②DMSA除轻度胃肠反应及出血倾向外,副反应较少,可作为长期维持用药。缺点为:Na-DMS出血倾向较重,易引致严重鼻衄及皮肤紫癜。
(三)二巯丙磺酸钠(Sodiumdimercaptosulphonate,DMPS)DMPS对重金属解毒作用与BAL相似,但毒性较BAL低约20倍,排铜效果远强于BAL。优点为:在各种排铜药物中,尿排铜量最高,副反应少。缺点为:偶见粒细胞缺乏症。
(四)D-青霉胺(penicillamine,PCA)PCA化学名为β,β-二甲半胱氨酸(β,β-dimethylcysteine),它是青霉素的水解产物,临床主要应用右旋青霉胺(D-penicillamine)和正-乙酰消旋青霉胺(N-acetyl-DL-penicillamine)。优点为:尿排铜增加达24.4μmol/24h,仅次于DMPS,而强于BAL、Na-DMS、DMSA及锌制剂等。缺点为:(1)副反应多,早期易发生过敏反应和白细胞减少,长期服药可发生SLE、MG、穿通性匐行弹性组织变性、粒细胞缺乏症及再生障碍性贫血等严重副反应;(2)长期服用,排铜作用逐渐衰减。因此,尽管国内外仍将PCA作为HLD的首选和常规治疗,但由于多种副反应,使需要终身服用排铜的HLD,往往被迫停药。因此,作者倡导多种排铜药中西医结合综合治疗为佳。
(五)依地酸二钠钙(calciumdisodiumedtate,CaNa2EDTA)口服吸收差,临床常采用肌内或静脉注射,于注射后1小时左右均匀地分布全身细胞外液,但不能进入红细胞内,药物属水溶性,故不易透过血-脑屏障,脑内浓度极低。优点为:价格低廉,副反应小,尿排铜高于BAL。缺点为:(1)因与锌、铁络合远高于铜;(2)连续使用,尿排铜作用渐减弱;(3)长期大剂量应用,可引起肾脏损害;(4)排锌、铁远高于排铜。
(六)三乙烯羟化四甲胺(triethylenetetraminedihydrochloride,trientine,TETA)TETA是一种多胺类金属络合剂,1982年美国食品与药物管理局(FDA)指定为对不能耐受PCA的HLD患者的治疗药物。本品极易吸收,迄今尚无有关TETA在体内代谢的研究报道。有人认为TETA在体内可能通过与球蛋白竞争和铜络合,使尿排铜增加。优点为:TETA排铜效果较高。缺点为:价格昂贵,可能致肾脏损害、EPS等严重副反应。
(七)锌制剂多数学者证明,食物中的锌抑制铜的吸收,血液中铜和锌的含量呈负相关,血浆锌浓度增高,铜含量相应减少。作者等(1986)对49例HLD口服硫酸锌治疗,36例于治疗3周后尿铜明显增高;1989年观察20例HLD口服葡萄糖酸锌,均于4周内尿排铜显著增高。表明锌制剂对体内贮积的铜有一定的清除作用。
wilson病_WILSON 氏病 -预后
本病除少数患儿出现酷似暴发性肝的临床表现,于短期内死亡外,其余自然病程大多呈缓慢进行性经过,约于起病后4~5年死于肝功能衰竭,或陷入植物人状态,死于肺炎等并发症。能生存10年以上者,极其少见,故预后不良。但在驱铜疗法进步的今天,绝大部分早~中期患者通过系统的中西医结合治疗,往往能获得与正常健康人相似的工作、学习和寿命。