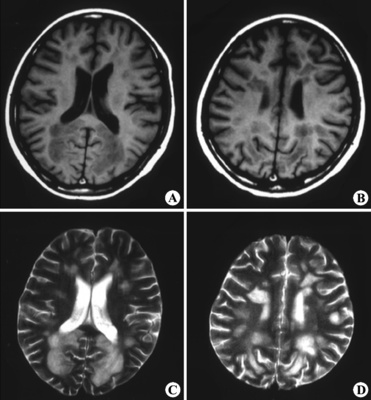
线粒体脑病,根据临床不同症候群可分为10型。
线粒体是人体重要的生产能量的细胞器,是人体细胞的主要能量来源。线粒体的基本功能是氧化可利用的底物,通过呼吸链电子传递合成ATP。因此,线粒体的结构和功能异常往往导致整个能量代谢过程紊乱,其中最主要症状是骨骼肌易疲劳,称为线粒体病。如病变以侵犯骨骼肌为主,称为线粒体肌病,如同时累及神经系统则称为线粒体脑病。 因受精卵中的线粒体均来自卵子,所以线粒体病系母系遗传,但也有散发病例。线粒体病多在青少年发病,表现为极度不能耐受疲劳,休息好转,常有肌肉酸痛和压痛,肌委缩少见。
线粒体脑肌病根据临床不同症候群又可分为10型。
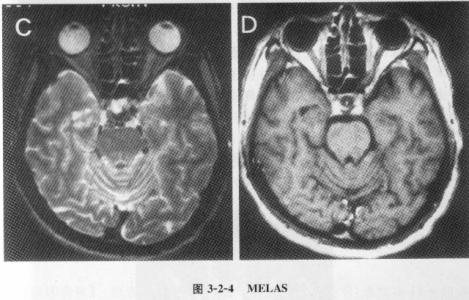
(1) MELAS综合征(为线粒体脑肌病、乳酸血症和卒中样发作):
临床特点为突发卒中,偏瘫、偏盲或皮质盲、癫痫发作、偏头痛和呕吐。病情逐渐加重,头颅CT和MRI显示枕叶软化灶,血和脑脊液乳酸增高。母系遗传,也可散发。
(2) MERRF综合征(肌阵挛性癫痫发作、小脑共济失调、乳酸血症和RRF):
主要特征为肌阵挛癫痫发作、小脑共济失调和四肢近端肌无力,多在儿童期发病。
(3) KSS综合征(视网膜色素变性、心脏传导阻滞和眼外肌麻痹):
20岁前发病、慢性进行性眼外肌瘫痪、视网膜色素变性,心脏传导阻滞、小脑共济失调、神经性耳聋、智能减退、脑脊液蛋白增高,病情进展快,多在20岁前死于心脏病。
(4)CPEO综合征:
各年龄均可发病,以儿童或成年早期发病为多,首发症状为眼睑下垂,缓慢发展为全眼外肌瘫痪、眼球运动障碍等。
(5) Leigh病(亚急性坏死性脑脊髓病):
于出生后6个月~2岁内发病典型症状为喂食困难共济失调,肌张力低下精神运动性癫痫发作以及脑干损伤所致的眼睑下垂,眼肌麻痹,视力下降和耳聋。临床上见到幼儿出现反复发作的共济失调肌张力降低手足徐动及呕吐症状应考虑此病。
(6) Alpers病(家族性原发性进行性灰质萎缩症):
(7) Menke病(卷毛型灰质营养不良):
(8) LHON综合征(Leber遗传性视神经病):
(9)NARP综合征(视网膜色素变性共济失调性周围神经病):
(10)wolfram综合征,主要表现为隐性遗传10岁起病,视神经萎缩,耳聋和早期发病的糖尿病,成年失明;
(11) MNGIE综合征(线粒体周围神经病并胃肠型脑病)。
线粒体病目前无特效治疗方法。可给予营养和支持治疗,延缓疾病的发展。词条图册更多图册