线粒体中的丙酮酸脱氢酶复合体(pyruvatedehydrogenaseComplex,PDC)能够催化丙酮酸氧化脱羧生成NADH和乙酰辅酶A,并把糖酵解与三羧酸循环以及ATP的生成紧密地联系在一起。这一重要反应对于需要大量ATP的组织(如大脑、肌肉)来说至关重要。通过PDC产生的乙酰辅酶A对于肝脏、脂肪这样能够产生脂肪的组织来说也是非常重要的,因为线粒体中的乙酰辅酶A可以通过形成柠檬酸(胞体乙酰辅酶A前体)进入到脂肪酸的合成过程。正是因为葡萄糖和脂肪酸之间存在这种底物竞争,所以高活性的PDC能够通过抑制肉毒碱棕榈酰基转移酶I来限制线粒体脂肪酸的摄取。由于在哺乳动物中不存在由乙酰辅酶A逆转生成葡萄糖的通路,因此在葡萄糖缺乏的时候抑制PDC的活性对于葡萄糖的转化至关重要。在肝糖元耗竭时,PDC反应的底物——丙酮酸作为葡萄糖合成的前体是肝脏和肾糖异生所必须的。这就使得血糖维持在一个稳定的水平,以保证不能够使用其他底物的组织获得ATP。除了脂肪酸氧化的产物(乙酰辅酶A和NADH)对PDC的异构抑制外,丙酮酸脱氢酶激酶(Pyruvatedehydrogenaseki— nase,PDK)通过磷酸化PDC 也可以抑制其活性.
PDC被PDK磷酸化失活后限制了丙酮酸被完全氧化或转变成脂肪酸。同时,通过抑制PDK来增加PDC的活性也是治疗糖尿病、心脏病以及肿瘤的药物靶点.
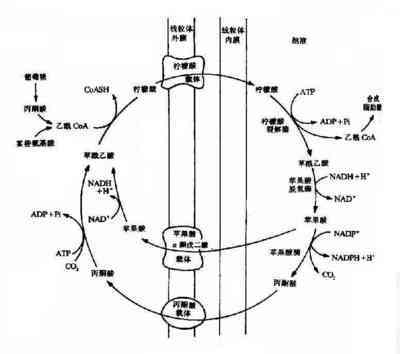
1PDK对PDC活性的调节
PDC是含有3个组分的复合体。这3个组分能够催化丙酮酸生成乙酰辅酶A:丙酮酸脱氢酶 (pyruvatedehydrogenase,E1)、二氢硫辛酰胺乙酰基转移酶(dihydrolipoamideacetyhransferase,E2)以及二氢硫辛酰胺脱氢酶(dihydrolipoamide dehydrogen-e,E3)。El催化生理可逆的步骤:PDK以及丙酮酸脱氢酶磷酸酶(pyruvate dehydrogenase phosphatephosphatases,PDP)调节的正是这一活性。PDK和PDP催化可逆的磷酸化(失活)和去磷酸化(激活)循环反应。El的磷酸化主要发生在3个特异的位点:264位丝氨酸(位点1)、271位丝氨酸(位点2)、203位丝氨酸(位点3)。位点1的磷酸化速度最快,而位点3的磷酸化速度最慢。因此,位点l的磷酸化是根据代谢需要快速调节活性PDC比例的主要位点.
在人类和啮齿类动物中目前鉴定出有4种PDK 同工酶:PDKI、PDK2、PDK3以及PDK4。其中PDKl在心脏、胰岛和骨骼肌中被检测到。而PDK2分布最广,在心脏、肝脏和肾脏中高水平表达。PDK3组织表达相对较局限(睾丸、肾脏和脑)。PDK4在心脏、氧化型肌肉、肝脏和肾脏等具有高度的脂肪酸氧化能力和高水平表达脂质氧化转录因子PPARd或者PPAR^y的组织中高丰度表达.
E1磷酸化位点突变分析表明,4种PDK都可以磷酸化位点1和位点2;而位点3只能被PDKl磷酸化⋯。对于位点2,PDK4比PDKI、PDK2和PDK3展现出更高的活性。PDK2可能通过磷酸化位点1来短期抑制PDC,而PDKl与PDK4对于多点磷酸化显得更为重要。如果PDK对于PDC的这种磷酸化调节异常,通常会引起一些人类疾病.
2PDK活性调节及自身的表达调控机制葡萄糖代谢产物能够高度调节PDK的活性.
乳酸(由糖酵解产生或是血液循环中)代谢产生的El的底物丙酮酸、NAD+以及辅酶A都对PDK活性起到抑制作用。而线粒体中PDC反应和脂肪酸B氧化的产物乙酰辅酶A及NADH能够激活PDK.
故而,增加线粒体中NADH/NAD+以及乙酰辅酶A/ 辅酶A的浓度比例可以增加PDK的活性.
营养条件和激素能够改变PDK的表达而影响葡萄糖一脂肪平衡。低血糖能够促使胰岛素的浓度降低从而使得脂肪分解增加,并最终导致脂肪酸氧化水平增高、葡萄糖的利用降低以维持血糖平衡.
实际上,脂肪代谢是经由过氧化物酶体增殖激活受体(peroxisome proliferator—activatedreceptor,PPARs)与基因表达信号通路相联系。PPAR信号通路能够促进组织中的PDK4在mRNA水平表达升高。在饥饿时的肝脏和心脏以及运动后的骨骼肌中,PPART共激活物Ot(PPARl/coactivatorOt,PGC—lot)表达水平迅速上调。在C2C12肌管中的研究表明,PGC.1仅和ERRa可以共同结合在PDK4基因的启动子区域并诱导PDK4的表达从而导致葡萄糖的氧化率降低。胰岛素在调节血糖中具有重要作用,它能促进葡萄糖氧化利用、抑制脂肪分解的作用。因此,饥饿诱导的胰岛素水平降低可以加剧脂肪组织中来源于三酰甘油的脂肪酸氧化代谢。给饥饿过夜的大鼠注入胰岛素5h后,骨骼肌中的PDK4在mRNA水平减少了72%,同时在蛋白质水平减少了20%,而胰岛素磷酸化Akt和FOX01则在其中发挥了重要作用。糖皮质激素也可以调节血糖。糖皮质激素通过位于转录起始位点上游820bp处的糖皮质激素反应元件与FOXOs结合在启动子的三个位点激活PDK4的表达,抑制PDC活性而升高血糖”.
3.1PDK与临床糖尿病机体在饥饿时主要由过表达PDK来抑制PDC的活性。
在糖尿病动物模型中也存在相同的调节机制来限制过量的葡萄糖消耗,这导致血糖和蛋白质糖基化水平升高,最终造成心血管系统的损伤。在肥胖个体中,葡萄糖的氧化水平也相应降低,这与胰岛素抵抗造成的活性形式PDC比例减少相关联,因而组织靶向控制PDK表达以及减少胰岛素抵抗个体肌肉中PDK4的表达具有潜在的治疗价值。已有文献报道应用DCA的研究,但是这一化合物是PDK的弱抑制剂,而且会产生毒性代谢产物。Novartis和AstraZeneca开发出一类新的抑制剂(Nov3r和AZD7545),这类抑制剂能结合到硫辛酰基基团结合位点,并有效地提高PDC的活性,故而能够有效地降低血糖。在肥胖大鼠的肝脏和骨骼肌中,PDK2与PDK4表达水平增加。抑制PDK2不但有益于肌肉组织(包括心肌)对葡萄糖的利用,更由于在肝脏中PDK的主要形式是PDK2,因此对于肝脏的葡萄糖利用也是有益的。选择性激活PDK2的另一个好处就是能够保留PDK4活性,而使得PDK4作为储蓄力量以预防在长期饥饿时血糖衰竭。由于肝脏PDC在脂肪酸生物合成中的重要作用以及PDK2是脂肪组织中主要的PDK形式,因此抑制PDK2能增强来源于葡萄糖的脂肪合成。AZD7545能特异性抑制PDK2,使得饥饿大鼠肌肉中PDC的活性增加了60%,而肝脏中PDC的活性增加了90%。显然,通过对PDK的干预激活PDC可以增加葡萄糖向脂肪酸的转化。因此,可能影响PDK表达的药物在糖尿病的治疗中具有极大的应用价值.
3.2心脏病
能量需求低时要降低PDC的活性,而在需要利用糖类以满足能量需求时要增强PDC活性,这对于心肌显得非常重要。有研究表明,在心肌局部缺血时,PDKl表达上调071,而通过抑制PDK来激活PDC已经被认为是治疗心肌病的有效途径。脂肪酸是心肌在中度局部缺血时主要的线粒体底物。在糖酵解时,利用脂肪酸有助于活性形PDC转化成非活性形式,使得丙酮酸转变成乳酸,并增加了心肌中的酸度。心肌细胞中的平衡态被打破,从而使得ATP减少以及钙离子摄取降低。目前DCA已经被作为心肌局部缺血时抑制PDK从而直接激活PDC的首要药物干预途径。在心肌局部缺血时,增加丙酮酸能够提高心脏在缺血时的机械运动。DCA能促进心肌预先使用丙酮酸而不是葡萄糖或者乳酸。在心肌缺血再灌注时,增加线粒体中钙离子的浓度也被认为是激活PDC的一个重要途径.
DCA有利于心肌局部缺血的治疗,其代谢毒性产物也带来了不良反应。但是,通过抑制PDK来激活PDC仍然是治疗心脏病的一个有前途的方向.
3.3癌症 在大多数肿瘤中都存在所谓的Warburg代谢.
与肌肉中的无氧酵解相类似,肿瘤中的葡萄糖也通过糖酵解转变成乳酸并分泌出来。在低氧化能力的实体瘤中,局部缺氧对于一些肿瘤中的这种代谢方式是一种合理的解释。然而在一些氧化能力较强的肿瘤细胞中,也同样存在Warburg代谢。无论是在有氧条件还是无氧条件下,肿瘤细胞对糖酵解的依赖与其恶性程度有关。最近的研究表明,迫使肿瘤细胞进行较多的有氧代谢能够抑制肿瘤的增长.
细胞要转变到Warburg代谢需要关闭PDC的反应.
在肿瘤细胞转变到Warburg代谢时,低氧诱导因子(hypoxia—inducingfactor,HIF)通路激活Ll。在肿瘤中,普遍存在能够直接或间接激活HIF信号的突变。HIF可以诱导PDKl的过表达而降低PDC的活性,最终维持乳酸的产生。因此,选择性地阻断HIF诱导的PDKl表达可以诱导肿瘤细胞的凋亡。已有报道称,抑制HIF或者PDKl可以有效地提高肿瘤细胞化疗的效果,并且这具有高度的肿瘤特异性。有研究表明,由于PDC活化造成的氧耗竭是肿瘤细胞凋亡的原因。还有研究认为,PDC的活化导致活性氧应激(reactiveoxygen spe—eies,ROS)增加并诱导肿瘤细胞的凋亡。不论是氧耗竭还是ROS的产生,都表明选择性激活PDC可以诱导肿瘤细胞的凋亡。因此,抑制PDKl或者其他形式的PDK都可以作为杀伤肿瘤细胞的潜在靶向.