做XRD有什么用途啊,能看出其纯度?还是能看出其中含有某种官能团?
X射线照射到物质上将产生散射。晶态物质对X射线产生的相干散射表现为衍射现象,即入射光束出射时光束没有被发散但方向被改变了而其波长保持不变的现象,这是晶态物质特有的现象。
绝大多数固态物质都是晶态或微晶态或准晶态物质,都能产生X射线衍射。晶体微观结构的特征是具有周期性的长程的有序结构。晶体的X射线衍射图是晶体微观结构立体场景的一种物理变换,包含了晶体结构的全部信息。用少量固体粉末或小块样品便可得到其X射线衍射图。
XRD(X射线衍射)是目前研究晶体结构(如原子或离子及其基团的种类和位置分布,晶胞形状和大小等)最有力的方法。
XRD特别适用于晶态物质的物相分析。晶态物质组成元素或基团如不相同或其结构有差异,它们的衍射谱图在衍射峰数目、角度位置、相对强度次序以至衍射峰的形状上就显现出差异。因此,通过样品的X射线衍射图与已知的晶态物质的X射线衍射谱图的对比分析便可以完成样品物相组成和结构的定性鉴定;通过对样品衍射强度数据的分析计算,可以完成样品物相组成的定量分析;
XRD还可以测定材料中晶粒的大小或其排布取向(材料的织构)...等等,应用面十分普遍、广泛。
目前XRD主要适用于无机物,对于有机物应用较少。
关于XRD的应用,在[技术资料]栏目下有介绍更详细的文章,不妨再深入看看。
如何由XRD图谱确定所做的样品是准晶结构?XRD图谱中非晶、准晶和晶体的结构怎么严格区分?
三者并无严格明晰的分界。
在衍射仪获得的XRD图谱上,如果样品是较好的"晶态"物质,图谱的特征是有若干或许多个一般是彼此独立的很窄的"尖峰"(其半高度处的2θ宽度在0.1°~0.2°左右,这一宽度可以视为由实验条件决定的晶体衍射峰的"最小宽度")。如果这些"峰"明显地变宽,则可以判定样品中的晶体的颗粒尺寸将小于300nm,可以称之为"微晶"。晶体的X射线衍射理论中有一个Scherrer公式,可以根据谱线变宽的量估算晶粒在该衍射方向上的厚度。
非晶质衍射图的特征是:在整个扫描角度范围内(从2θ1°~2°开始到几十度)只观察到被散射的X射线强度的平缓的变化,其间可能有一到几个最大值;开始处因为接近直射光束强度较大,随着角度的增加强度迅速下降,到高角度强度慢慢地趋向仪器的本底值。从Scherrer公式的观点看,这个现象可以视为由于晶粒极限地细小下去而导致晶体的衍射峰极大地宽化、相互重叠而模糊化的结果。晶粒细碎化的极限就是只剩下原子或离子这些粒子间的"近程有序"了,这就是我们所设想的"非晶质"微观结构的场景。非晶质衍射图上的一个最大值相对应的是该非晶质中一种常发生的粒子间距离。
介于这两种典型之间而偏一些"非晶质"的过渡情况便是"准晶"态了。
在做X射线衍射时,如果用不同的靶,例如用铜靶或者Cr靶,两者的谱图会一样吗?如果不同的话,峰的位置和强度有啥变化吗?有规律吗?
不同的靶,其特征波长不同。衍射角(又常称为Bragg角或2θ角)决定于实验使用的波长(Bragg方程)。使用不同的靶也就是所用的X射线的波长不同,根据Bragg方程,某一间距为d的晶面族其衍射角将不同,各间距值的晶面族的衍射角将表现出有规律的改变。因此,使用不同靶材的X射线管所得到的衍射图上的衍射峰的位置是不相同的,衍射峰位置的变化是有规律的。
而一种晶体自有的一套d值是其结构固有的、可以作为该晶体物质的标志性参数。因此,不管使用何种靶材的X射线管,从所得到的衍射图获得的某样品的一套d值,与靶材无关。
衍射图上衍射峰间的相对强度主要决定于晶体的结构,但是由于样品的吸收性质也和入射线的波长有关。因此同一样品用不同靶所取得的图谱上衍射峰间的相对强度会稍有差别,与靶材有关。
重温一下布拉格公式和衍射的强度公式,您的问题答案全都有了。
我想知道不同衍射角对应的晶面,怎么办?
如果你的图能够找到对应的粉末衍射数据卡,那么问题就简单了。多数的粉末衍射数据卡上面都给出了各衍射线的衍射指标,也就可以知道对应的晶面了。
如果是未知晶体结构的图,就需要求解各衍射线的衍射指标,这一步工作叫做"衍射图的指标化"。如自己解决需要具备基础的晶体学知识,然后学会一两个指标化的工具软件(如treaor90)进行尝试。
对于正交晶系的晶胞参数,其中a、b、c代表晶胞的三个棱的长度。但我不清楚如何定义a、b、c的方向,也就是说按照什么依据确定这三条棱的方向?是否有明确的规定还是可以任意自定义?
一般来说可以用a < b <c的定向原则,其实,用什么方向都可以,它们可以通过矩阵来转换。
晶胞中的a,b,c,分别是三个晶轴方向上的单位平移向量的长度,称为轴长,不是"三个棱"的长度。轴长符号也常用a0,b0,c0表示。轴长单位常用?(埃,Angstrom,=10-10米)或纳米(nm,=10-9米)。在晶体结构中没有"棱"这样一种说法,只有晶体坐标系,而这个坐标系是用a,b,c,α,β,γ 六个参数来表示的,α,β,γ分别代表三个轴间的夹角。而"晶棱"是指晶体的外形的棱边。所以说"a、b、c代表晶胞的三个棱的长度"是错误的。
如何计算晶胞体积?比如说我想计算二氧化锆四方晶相的晶胞体积,甚至是各个晶胞参数,怎么用这个软件来具体处理一下呢?
首先,你要有相应的晶体学方面的知识。这些软件是为我们处理一些晶体学上的一些问题服务,所以,你不能抛开晶体学去使用软件。有了一些必要的晶体学知识之后,你再去学习使用这些软件,这样你才能看懂help里的内容。对于你现在所讲的这个晶胞体积的问题,实际上也就是晶胞参数精确测定的问题,因为晶胞参数精确测定了之后,晶胞体积自然就知道了。
有什么软件能根据分数坐标画出晶体的空间结构?就是有八面体或者四面体的那种。
根据晶体的结构结构数据,用diamond或atoms等专业的晶体结构绘图软件便可画出晶体的空间结构。
六角结构的晶体在生长时它的内在的优先生长方向是哪一个?
一般来说晶体沿短轴方向生长速度快,垂直于长轴方向的晶面密度较大,从能量的角度说,当晶体生长时,这样的格位更稳定一些。
在X射线测量中(三方晶系)通常给出的都是六方的晶格常数例如a=b=5.741,c=7.141,夹角分别为120°,120°,90°。现在我想把它换算成三方结构的晶格常数a=b=c=? , 夹角 a=?
你是通过对衍射数据指标化得出的六方晶格常数,还是从文献得到的?如果你的晶胞是菱方格子,那么用六方定向和菱方定向是一样的
梁敬魁的《粉末衍射法测定晶体结构》中有公式可以由三方转六方,或六方转三方。
如何知道晶体中原子坐标?
做单晶X-射线衍射才能得到原子的坐标。除了四圆外,CCD也可进行单晶X-射线衍射。
如何根据X射线衍射数据计算晶粒尺寸晶格常数和畸变,用什么理论和公式?
根据衍射峰的峰形数据可以计算晶粒尺寸晶格常数和畸变。在衍射峰的宽化仅由于晶粒的细小产生的情况下,根据衍射峰的宽化量应用Scherrer公式便可以估算晶粒在该衍射方向上的厚度。你如果需要做这方面的计算,需要增加一些入门知识,在本网页上你就能够找到一些有关资料的。
[X射线小角衍射和X射线小角散射]
小角X射线散射(Small Angle X-rayScattering)和小角X射线衍射(Small angle x-ray diffraction)是一回事吗?
早期小角X射线散射仅指超细颗粒在低角度范围(常指2θ<20°)上的X射线散射,而现在,小角X射线散射通指在低角度范围(常指2θ<10°~20°)的X射线散射。
X-射线照射到晶体上发生相干散射(存在位相关系)的物理现象叫衍射,即使发生在低角度也是衍射。例如,某相的d值为31.5A,相应衍射角为2.80°(Cu-Kα),如果该相有很高的结晶度,31.5A峰还是十分尖锐的。薄膜也能产生取决于薄膜厚度与薄膜微观结构的、集中在小角范围内的X射线衍射。在这些情况下,样品的小角X射线散射强度主要来自样品的衍射,称之为小角X射线衍射。对这类样品,人们关心的是其最大的d值或者是薄膜厚度与结构,必须研究其小角X射线衍射。
X-射线照射到超细粉末颗粒(粒径小于几百埃,不管其是晶体还是非晶体)也会发生相干散射现象,也发生在低角度区。但是由微细颗粒产生的相干散射图的特征与上述的由超大晶面间距或薄膜产生的小角X射线衍射图的特征完全不同。
小角衍射,一般应用于测定超大晶面间距或薄膜厚度以及薄膜的微观周期结构、周期排列的孔分布等问题;小角散射则是应用于测定超细粉体或疏松多孔材料孔分布的有关性质。
X-射线照射到样品上还会发生非相干散射,其强度分别也主要集中在在低角度范围,康普顿散射就属于此类,其结果是增加背景。
哪里能得到小角X射线衍射的系统理论包括书、文献、技术、软件?
1. 张晉远等, X 射线小角散射. 高等教育出版社, 北京, 1990
2. Y. Xiang, et al. Materials Characterization,2000, 44(4-5): 435-9
我现在做介孔材料。介孔(孔径2-50nm)在材料中成有序排列,象晶格一样的排列在材料中,孔壁、材料为非晶相。为什么XRD能粗测孔与孔的间距?我了解到的是,孔成有序排列,所以在小角度会有衍射峰,(001)面的峰值和孔径有关。但我不知道为什么?
跟长周期有关:大的孔需要大的周期,或者说是"孔面"间距,类似于"晶面间距"。"孔"意味晶体中该区域没有原子填充,没有填充原子就无衍射峰。而孔洞的边界是原子紧密排列的,原子密度相对较高,对应产生较强的衍射,强度较大。大孔孔径大,空间重复周期大(即长周期),对应的晶面距大,产生的衍射在小角区。所说的(001)有强线对应的材料的晶体C轴较长,如果第一线是(100)则A轴较长。
[关于粉末衍射数据库的问题]
PDF2卡片与JCPDS 卡有什么区别?
是同一个东西!
PDF2是ICDD (International Centre for DiffractionData)的产品,ICDD的前身为JCPDS (Joint Committee on Poder DiffractionStandards) 。
为什么我用pcpdfwin查到的电子PDF卡和我在学校图书馆查的PDF卡不同,卡号都是一样的,可相对强度值不一样,这是为什么?
相对强度是估计的,有误差是正常的,可能是数据的来源有所不同造成的,还有要注意,由于卡片制作后就不能改,所以有的卡片被后来的结果修正了,这在印制的卡片上是没有这种信息的,但在数据库中的卡片则有说明,哪些卡片已经被删除。
衍射卡片里面相对强度怎么有的大于100?(比如为999)
粉末衍射卡的强度数据以相对强度提供,一般以最强线为100。但是计算的粉末衍射数据最强峰的强度值取作999。其实相对强度的数值并不重要,您只要把最大的当作100,其它的与之比较就可以了,比如999当作100那么500就是50,353就是35,在这里因为是http://jf.chinamobile.com估计,有误差也没有关系了。
同一种物质对应着两张卡片,这正常吗?
这很正常,两张卡片是在不同的时间或由不同的人做的。你可以按卡片号调出卡片,卡片上就可以查到卡片数据出处的原始文献。
在X射线粉末衍射的数据表中,Peak List中有Rel.Int.[%],PatternList中有Scale Fac,请问"Rel.Int.[%]","Scale Fac"代表的意义。
"Rel.Int.[%]"的意思是"相对强度,%","ScaleFac"是(强度)"比例因子"。
请问哪里能查到文献上发表的天然产物的晶体数据?
ICSD 数据库或ICDD 的PDF数据库。
除此之外,还可以在矿物数据库中和美国矿物学家晶体结构数据库中免费查到,在"晶体学数据库"栏目中提供的链接中查。
[物相分析方面的问题]
样品卡与标准卡对比原则?
提供几条原则供大家参考:
1. .对比d值比对比强度要重要;
2. 低角度的线要比高角度的线要重要;
3. 强线比弱线重要;
4. 要重视特征线;
5. 同一个物相可能有多套衍射数据,但要注意有的数据是被删除的;
6. 个别低角度线出现缺失;
7. 由于样品择优取向某些线的强度会发生变化;
8. 有时会出现无法解释的弱线,这是正常的,不能要求把所有的线都得到解释。
"要重视特征线",那么什么是特征线?"同一个物相可能有多套衍射数据,但要注意有的数据是被删除的",这是什么意思?
所谓的特征线就是某物质最强而且是独有的最容易判断的线,如石英的特征线就是d=3.34?的线,在混合物中如果出现这条线,有石英的可能性就大,其它也是这样,这在混合物中查物相是很有用的。
同一个物相可能有多套衍射数据是指有多个卡片的数据都是一个物相,比如石英(SiO2)从1到49卷都有数据,共有93个数据卡片,但是1-8卷15,16,33-1161,42-391等(共38个)这些卡片都是卡片库的编者已经删除的。
哪里能查到文献上发表的天然产物的晶体数据?
除ICSD database 和ICDD的PDF库外,还可以在矿物数据库中和美国矿物学家晶体结构数据库中免费查到。
如何将单晶数据转化为粉末的,来作为我的标准谱呢?
可以利用Shape公司的软件,利用单晶数据计算粉末理论图,最新版本1.2demo版。
没有它的粉晶射线图,但是有根据其单晶X射线衍射推算出来的晶胞参数,请问我怎么才能反推算出该晶体的粉晶衍射图?
如果你有结构数据,就可以从理论上算出粉末衍射图的,可以使用MaterialsStudio的reflex plus模块算出来;有原子坐标,通过diamond也可以得到它的粉末XRD图。
如何分析出其晶体结构而确定是我们预期的物质呢?
如果能够在ICDD的粉末衍射数据库中找到你预期的物质的衍射数据,做一个XRD鉴定即可确定。
做了一些铝合金试样的XRD,试样中铝含量超过95%。试样的谱线均在纯铝谱线左侧,低角度谱线偏离0.2度左右,高角度谱线偏离0.3-0.4度左右。请问它们是第二相固溶引起的吗?它们与固溶程度有什么定性和定量的关系?
根据Bragg公式,峰位置向左偏移,相当于d值变大了,反映其晶胞参数变大了,说明在Al的晶格中渗入了其他的原子。另外,没有出现新的衍射峰,说明是铝的无序固溶体,保持着纯铝的晶体结构。
钴酸锂的标准卡片有哪几种?而许多厂家用的是16-427标准卡片,没有考虑(009)这个衍射峰,为什么不用75-0532标准卡片呢?
16-427是通过实验得出的,而75-0532是利用ICSD结构数据库中的结构数据计算得出的
做钴酸锂实验时,出现的峰和75-0532一样,而和16-427是不是只差(009)衍射峰呢?如果是这样,那么就是16-427漏掉了这个峰,或者当时实验者收集数据时的角度不够高,没有达到这个峰的位置,这种情况是常有的
做钴酸锂XRD衍射实验时,在X衍射仪上做出的衍射图片,第一个衍射峰特别高,其他的衍射峰特别低,不成比例,是仪器本身的原因,还是发生了择优取向,请各位高手指点。
不知你是用钴酸锂粉末做的实验,还是多晶膜等?如果是粉末,那可能就是择优取向了
化学沉淀法从氨氮废水中结晶出磷酸铵镁,XRD与标准JCPDS相比衍射峰有规律向左偏移,这表达什么晶体结构的信息啊?
可能的原因:
如果各峰的偏移基本上是一个固定值,原因是2θ零位不正确;
样品平面后仰;
组成偏离化学式"MgNH4PO4·6H2O",未知的偏离导致晶格变大了一些。
用Jade5软件可以进行XRD定量分析吗?
XRD定量分析的基础数据是样品中各组成物相的强度数据(原理上应该用峰的面积数据),余下的工作便是些乘除比例的运算了,使用表格软件(如WPSOfice的"WPS表格"、微软的Excel)完成甚为方便。因此,使用任一能够获得峰面积或峰高的软件工具都可以进行XRD定量分析,当然,有Jade软件更为方便。
哪里有X射线衍射校正和定量分析用的标准物质(GBW(E)130017)α-石英(α-SiO2)
国家标准物质研究中心询购。http://www.nrccrm.org.cn
[结构分析计算方面的问题]
请介绍一些介绍Rietveld方法的书籍。
这方面的书籍很多,如:
《粉末衍射法测定晶体结构》
作者:梁敬魁编著 出版社:科学出版社
出版时间:2003 丛编项:应用物理学丛书
简介:本书系统全面论述了粉末衍射图谱的指标化,点阵常数的精确测量,粉末衍射测定新型化合物晶体结构的各种方法等在离子晶体结构分析中的应用。
主题项:粉末衍射法-晶体结构 X射线衍射-晶体结构
有没有关于精修的详细操作的指南?
如果你想用Fullprof来精修结构,那么你可以看看本论坛:http://www.crystalstar.org/Article/ShowArticle.asp?ArticleID=81关于这个软件的使用说明。
有了结构数据后,你可以用diamond、atoms等绘图软件绘出晶体结构图
请推荐一个小巧玲珑,方便实用的计算晶胞参数的软件。
Celref2.0或3.0都可以。
chekcell软件可以根据xrd图谱和pdf卡片获得样品的晶胞参数么?
CHEKCELL是进行晶胞参数的精修,粗晶胞当然可以在pdf卡片上得到了,这个软件是较简单,按照上面的提示操作就可以了
用XRD图来精修出分子结构研究分子的性质, 收集XRD时应注意些什么?
强度要高,中等强度的衍射峰强度要达到5000计数以上;衍射峰的分辨要尽可能的好;扫描范围要大,最大d值的峰不能缺失。衍射峰的强度和很多因素有关,比如样品的衍射能力,性质,还有仪器功率,测试方法,检测器的灵敏度等等。
做XRD时步长一般为0.02度,但是如果要做Rietveld分析,出了强度要求500以上,步长有没有什么要求啊?
一个半峰宽内有3-5个点就可以了,步长一般为FWHM的0.2-0.3就可以了。
强度要求在10000-20000之间。
如果用普通的xrd仪器如brucker D8 或者 philips等测试粉末的xrd,扫描速度需要多慢才适合用gsas等方法作rietveld分析,需要加内标吗?
扫描速度的快慢是根据你的数据强度要求来确定的,如果要做结构精修,中等强度的衍射峰的强度应该在5000以上,至于内标是不需要的。
衍射峰的强度好像和仪器有很大的关系,因为以前用Rigaku的Dmax,每次都是几千,但现在用Bruker或者Philips,好像每次都在一千以下,不知道是什么问题。粉末的结晶肯定是好的。
与发生器的功率有关。你曾使用的Rigaku的Dmax是高功率的转靶衍射仪吧?强度也和衍射仪的扫描半径有关。
用国产仪器(丹东方园仪器有限公司)测的XRD数据能不能用于Retrieve精修?
国产衍射仪的基本技术指标(测角准确度、重现性、分辨率,衍射强度稳定性、长期工作稳定性等指标)与进口衍射仪并无水平级的差异。配有石墨弯晶单色器的国产衍射仪,正确选择狭缝、采数步宽和步进计数时间(保证峰强度),同样能够获得能够用于Retrieve精修的衍射数据。例如中科院北京研究生院无机新材料实验室近几年来完成的大量晶体结构分析工作,其衍射数据就是在该实验室购置的XD-3型X射线衍射仪(北京普析通用仪器公司生产)上收集的。
我做的是磷矿粉的XRD,它属于六方晶系。请问有没有能计算晶胞参数的程序,以及程序的用法。
首先您应该要求给您作衍射的实验室提供d值和强度的衍射数据,然后检索ICDD卡片。如果你把这个物相的卡片数据检索到了,那么你的问题就是晶胞参数精修了,下载Chekcell精修晶胞参数。如果你还想计算结晶粒度的大小,建议你用一些全谱拟合的软件,如Fullprof等,这样,不仅可以精修晶胞参数,同时也可以得到半高宽或积分宽度的数据,可以用于结晶粒度计算。当然,要计算结晶粒度,最好还要进行仪器校正曲线的测定,一般在收集你的实验样品的同时,再在相同条件下收集标准样品(LaB6,标准Si等)的谱图,以便扣除仪器对半高宽或积分宽度的贡献。
已经知道晶体的晶格常数、晶系以及各衍射峰的hkl值,请问如何确定其空间群,有没有相应的软件。
有了这些就可以用Shelx来解晶体了,空间群自然也就能确定了。可以用国际晶体学手册,利用你的已知数据便可查的。未知物质的空间群是根据统计消光规律来得到的,最终的结果还要用结构测定的结果来最后确定,统计消光规律来推测空间群的软件有XPREP,wingx等.
具体可以如下操作:衍射数据当作P1空间群处理然后利用 Pattern 程序生成Psudo-Precession然后根据系统消光规律推导出晶体的劳埃对称性。不是所有的时候都可以直接给出最终的空间群。最好在解完结构之后,利用结构解析程序产生的FCF文件再重新核对一遍。
如果我想要從這個 powder diffractionprofile來解出其結晶結構.包括其各個晶格參數 以及其是什麼樣的晶型 tetragonal or orthorhombic等等那我該如何是好呢?
如果你只想知道这个新物相的晶格参数和晶系,用你的粉末衍射数据去指标化就可以了。指标化就是确定每个衍射峰的衍射指标,当然这个过程同时就给出了晶系。
有沒有一個程式是可以輸入 a b c三軸的值以及三軸之間的三個夾角然後就可以自動畫出這樣的晶型可能產生的 powder diffractionprofile呢?
有这样的程序可以输入晶格参数和空间群就可以理论计算出衍射峰的位置。可以用的软体很多,CHEKCELL这个免费的软体就可以解决您的问题。如果需要理论计算衍射强度,你还需要给出原子坐标参数。
原子各向同性(异性)温度因子对结构影响大吗?
温度因子就是反映原子在平衡位置附近振动情况的一个因子,主要是对强度有影响,那当然就对结构有影响了。
晶体的各向异性温度因子是如何定义的?
晶体中的原子普遍存在热运动,这种运动在绝对零度时也未必停止。通常所谓的原子坐标是指它们在不断振动中的平衡位置。随着温度的升高,其振动的振幅增大。这种振动的存在增大了原子散射波的位相差,影响了原子的散射能力,即衍射强度。在晶体中,特别是对称性低的晶体,原子各个方向的环境并不相同,因此严格的说不同方向的振幅是不等的,由此引入了各向异性温度因子。
在进行Rietveld结构精修时,是否该对温度因子进行约束?如何约束以及约束范围?
由于温度因子是随着衍射角的增加而对强度的影响增大,所以,如果要精修温度因子,就一定要收集高角度的数据。
如何由粉末衍射数据通过FullProf提取结构因子?
首先需要一个dat文件,第一行,2theta起点,步长,终点,下面是每个点的强度。
下面需要编写pcr文件,先得到六个晶胞参数,零点,还要得到18-30个背景点,才能开始编写,其他参数设置可以看说明书。如果你以前用Fullprof精修过结构,则只需修改如下参数:Line 11-2 的N(number of atoms in asymmetricunit)参数置为0,相应的下面与原子有关的参数就不要了;Line 11-2的JBT参数(2,-2,3或-3,具体看说明)。至于要输出什么样格式的结构因子数据文件,可以通过LINE 3的JFOU参数来控制。
Fullprof精修时,Biso的值给如何设定?是否有个大概的取值范围?
Biso是温度因子,occu是占有率。从我拟合来看,Biso与原子的位置有关系。温度因子是反映原子或离子偏离平衡位置的程度,因为晶胞中各原子都要做热振动的。对于立方晶系,各向同性,只修各向同性温度因子就可以了。温度因子和占有率都是影响强度的参数,所以之间有一定的相关性。而且,温度因子对高角度峰的强度影响比较大,所以,如果要精修温度因子,最好收到高角度的数据。
Fullprof精修中的scale的初值一般是怎么确定的?
比例因子scale是指理论计算数据与实验数据之间的比值,一般随便给就可以,不会影响精修的。只要你的初始模型正确,几轮就可以修到比较合理的值。
我最近才接触到Rietveld精修,使用的软件是Fullprof。在编写pcr文件的时候碰到一些问题,比如:ATZ跟OCC这两个参数是怎么算出来的。我查了一些资料,计算的公式是有了,但是具体的那些参数的值是怎么算的就不清楚了,还请各位多多指教!
AZT = Z.Mw.f^2/t
Occ= chemical occupancy x sitemultiplicity (canbe normalized to the multiplicityof the general position of thegroup).
请问:1. Z,f,t,该怎么取值?
2. 什么是general position of thegroup,从哪里可以获得这个数据
答:
1. AZT = Z*Mw*f^2/t
(useful to calculate the weight percentage ofthe phase)
Z: Number of formula units per cell
Mw:molecular wheight
f: Used to transform the site multiplicitiesto their true values. For a stoichiometric phase f=1 if thesemultiplicities are calculated by dividing the Wyckoff multiplicitym of the site by the general multiplicity M. Otherwise f=Occ.M/m,where Occ. is occupation number.
t: Is the Brindley coefficient that accountsfor microabsorption effects. It is required for quantitative phaseanalysis only. When phases have like absorption (in most neutronuses), this factor is nearly 1. If IMORE=1 the Brindley-coeff isdirectly read in the next line (in such case ATZ=Z.Mw.f^2).
这里说明一下,ATZ的值只有在做定量分析的时候有用,如果不做定量分析,只做结构精修,则可以随便给一个值即可。
2. general position of thegroup就是晶体学国际表中某个空间群的一般等效点系数,可以从晶体学国际表查得:International Tables forCrystallography, Volume A: Space GroupSymmetry
刚开始学fullrpo,我样品里可能有二相,空间群为C2/c(主相)和R-3c,我想得到各自的结构信息,先修scale-zero-background(前三参数)-UVW-cellparam.几轮下来来峰位大致对上,可是再修Shape1误差就更好,修原子坐标也很离奇,出现2点多和负值。
请问:
1,原子坐标修的顺序怎么定呀,我看程序附带的eg.里修原子的顺序不是按原子顺便排下来的?
2,ATZ值再我这重要吗?手册里公式ATZ=ZMf^2/t中个系数分别什么意思呀?
3,原子占位=chemOCC.m/M中个系数又是什么意思呢?
4,原子各向同性(异性)温度因子Biso/B11,B22,B33,B12,B13,B23对结构影响大吗?他们是什么意思呢?
答:
修原子坐标时,一般是由重到轻,也就是先修原子序数大的;
ATZ公式里各个系数在fullprof的帮助文件里说的比较清楚;
原子占有率中m是特殊等效点系的重复数,M是一般等效点系的重复数;
温度因子就是反映原子在平衡位置附近振动情况的一个因子,主要是对强度有影响,那当然就对结构有影响了,你可以看看结构因子的表达式,就可以找出到底有啥影响了。
要做原子的占位及占有率。Z、t的值都可以随便取吗?Z是表示晶包中原子的个数吗?
随便给一个ATZ的值就可以了。Z是表示晶胞中所包含的化学式的个数。
就是在用Fullprof这个软件精修晶格参数时,对于几个R因子,Rp,Rwp,Rexp等参数的值大小一般要为多大精修的结果才可靠?
Rp,Rwp参数的值大小通常和你的数据质量、结构模型等有关,目前好像没人说这个值应该是多少。Rexp是一个期望的因子,它与数据点数、精修参数的个数等有关。对于晶胞参数,通常主要看R-Bragg因子。对于这些因子你可以看看这篇文献:J.Res. Natl. Inst. Stand. Technol. 109, 107-123 (2004)
用fullprof软件做rietveld 精修一般要做很多轮,而且在过程中要不断检查和调整PCR文件,我想请问一下,像温度因子(要求不能为负数的),占有率,各项异性等在那个文件里看啊?
算完后直接在.new(如pcr=2)或.pcr(如pcr=1)里就能看到的。
使用Fullprof软件进行Rietveld精修时,编辑PCR文件过程中,Nex参数的含?什么情况下使用Nex?Nex的数值选0、1、2的依据是什么?排除区域的范围如何确定?
Nex就是排除区域的个数,即Nex=0时,没有排除区域;Nex=1时,表示有一个排除区域,依此类推。被排除的区域是不参加拟和精修的。
所有JCPDS卡片上的物质,都可以从ICSD数据库输出*.cif文件吗?
JCPDS是粉末衍射数据库,而ICSD是无机晶体结构数据库,所以对于JCPDS卡片中的某一物相,只要ICSD数据库中有对应的物相,就可以从ICSD数据库中导出*.cif文件。但并不是所有的JCPDS卡片中物相都能在ICSD数据库中找到对应的结构数据,因为我们知道,对于某一物相,我们要收集到它的粉末衍射数据是比较容易的,但是,对于得不到单晶的物相来说,要想从粉末数据解出晶体结构是不容易的。
如何由XRD的数据和峰值得出diamond画图需要的cif文件,请问需要什么软件
由XRD的数据通过一系列分析(如寻峰、指标化、提取结构因子、解结构等步骤)得出结构参数,然后才能得到cif文件。
若结构已知,则可以直接从Icsd数据库输出cif文件
如何将TXT转化为CIF格式?衍射仪能否将数据直接存为CIF格式?"将*.txt文件用记事本打开,文件名另存为*.cif格式,其中的保存类型选项为"所有文件",保存"这样做对吗?
* .cif文件是在解好结构以后,用acta指令产生的。测晶体后只能得到强度文件和设置文件。而不是把文件扩展名txt直接修改为cif。
如何将粉末衍射的dat数据转成用于expo的pow数据,是用什么软件还是直接手改的。用什么软件可以生成后缀为pow的文件。
把数据导入PowderX,输出数据为.pow就可以了。PowderX的数据格式可设定为:
abc /注释行/
数据点总数 /(终止角度-起始角度)/步长+1/
强度 2Theta
... ...
... ...
存为.dat文件,然后导入数据,选择x-y (.xrd)。
当然有一些知名公司仪器的格式可以直接导进去。例如可以用Jade ,如果你知道*.pow文件的格式。
做Rietveld时发现:如果事先对原始数据进行平滑等一系列的预处理会比较容易使R因子减小,小弟的做法是否不妥?
做Rietveld时,是不需要对数据进行平滑处理的,因为平滑处理肯定会使数据在一定程度上失真的。不过,在不影响峰形的前提下进行轻度的平滑处理也是可以的
在做Rietveld精修(refinement)之前我已经精确的测定了晶胞参数。在精修的过程中,发现晶胞参数与先前的测定值相比较,差别很大,但整体的R残差因子(R-factor)很小。 如果对晶胞参数进行束缚,则整体的残差因子较大。请问我该如何处理?
如果你的XRD数据没有杂相峰,且提供的空间群也正确,那么应该优先选择最小的R值的结果。
我下载安装了Fullprof软件,但打不开在理学DMAX-2000仪器上的数据文件.raw,或.txt文件。请问它要求的文件格式是什么吗?该怎么转化成它要求的文件格式?Fullprof和winplotr两个软件哪个好用?
你可以用jade将数据转换为Fullprof能识别的格式,Fullprof能识别多种数据格式,具体格式在它的说明文件里都有相应的说明。
Fullprof是一个可单独执行的用于晶体结构分析的程序;而Winplotr是一个用户界面程序,我们通过它来调用Fullprof、指标化程序、键长键角计算等程序
如何计算出来理论衍射图(给定晶格参数和实验参数后,如何计算得到X射线衍射图)?
首先,模拟(就是你说的"计算")是要基于许多理论模型的,因为粉末衍射图样的形成要受很多因素的影响,峰的位置、形状,峰彼此之间的影响都可以用一些模型来描述,三两句话说不清楚;
第二,有大量的现成的软件可以用;
第三,如果你打算把粉末衍射的模拟作为你的研究课题,你应该多看一些文献和基本原理的介绍。只要你有了晶胞参数、原子坐标,就可以用大量的现成的软件来计算出仿真的X射线粉末衍射图。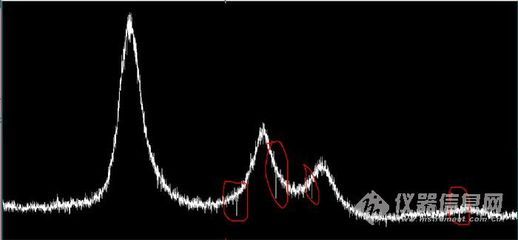
有单晶X射线衍射推算出来的晶胞参数,请问我怎么才能反推算出该晶体的粉晶衍射图?
需要原子坐标!如果你有结构数据,就可以从理论上算出粉末衍射图。例如,通过Fullprof、Diamond软件工具即可以得到该晶体的粉末XRD图,也可以使用Materials Studio的reflexplus模块算出来,不用自己手工算了。
请问知道晶体的结构和晶格参数能模拟它的X衍射谱线吗?
这种软件很多,crysconI.exe就是其中一个。用PCW也不错,可以转化成txt文件,在origin画图进行比较,很方便
[峰宽问题]
除了结构缺陷和应力等因素外,为什么粒径越小,衍射峰越宽?
从衍射理论知道,衍射极大和第一极小之间的角宽度与发生相干衍射区域(相干域)的尺寸有关,相干域越大,角宽度就越小。一般来说,相干域的尺寸小于2微米,就会使衍射峰造成可测量的宽化。所以,晶粒的粒径越小,以至不能再近似看成具有无限多晶面的理想晶体,对X 射线的弥散现象就越严重,表现在峰强变弱,峰变宽。
什么是标准半峰宽度,如何得到?
所谓的标准半峰宽应该是指仪器本身的宽化因子,和实验时使用的狭缝条件关系最大,想得到它并不难:
比如在相同的测量条件下,把Si标样放到仪器上测量Si的各个衍射峰的Kα1峰的半高宽,就是所谓的标准了。当你需要测量一系列非标样Si粉时,就把标样Si的Kα1峰的半宽作为标准半峰宽使用就可以了。
但,有很多时候合适的标准物质很难得到,你就用另外的标准物质(出峰位置很相近的标准物质)代替,也完全没有问题。也可以根据Si标样在整个扫描范围内的衍射峰的Kα1峰的半高宽作出仪器宽化因子-2θ关系曲线来得到任意进度的仪器宽化因子。因为谢乐方程的适用条件也就是几十到200nm之间,超出这个范围误差是很大的。只要你在进行相同的一系列计算时使用相同的一个参数就一般就可以满足研究工作的要求了。
为什么晶粒尺寸的变化会引起X射线衍射的峰线宽化?
理想的晶体是在三维空间中无限的周期性延伸的,所以,如果不考虑仪器宽化的因素,那么理想晶体的衍射峰应该是一条线,但是实际晶体都是有尺寸的,即,周期性不是无限的,这就造成了由于结晶粒度引起的宽化,如果结晶粒度无限小下去,衍射峰就会宽化直至消失变成大鼓包了,也就是非晶了。
晶体晶粒细化,多晶试样中存在宏观应力时,衍射花样的变化情况是怎样?
不管是粉末试样还是(多)晶体试样,粉末颗粒或晶粒太粗,参加衍射的晶粒少,会使衍射线条起麻,衍射的重现性但粉末颗粒或晶粒过细时,会使衍射线条变宽,这些都不利于分析工作。存在宏观内应力的效应是使得衍射环或衍射峰的位置改变,导致底片上的衍射线条变宽,不利于分析工作。
[衍射图数据收集方面的问题]
收集XRD应注意些什么?(比如收集角度泛围、速度等有什么要求?
衍射峰的强度和很多因素有关,比如样品的衍射能力,性质,还有仪器功率,测试方法,检测器的灵敏度等等。
XRD衍射强度和峰的宽度与样品颗粒大小,还是与晶体颗粒大小有关?
样品中晶粒越小,衍射峰的峰高强度越来越低,但是峰越来越宽,实际上利用X射线衍射峰的宽化对样品的结晶颗粒度分析就是根据这个原理的(Scherrer公式)。
晶粒大小和颗粒大小有关系,但是其各自的含义是有区别的。一颗晶粒也可能就是一颗颗粒,但是更可能的情况是晶粒抱到一起 , 二次聚集 ,成为颗粒。颗粒不是衍射的基本单位噻, 但是微小的颗粒能产生散射。你磨的越细 , 散射就越强。对于晶粒, 你磨过头了,晶体结构被破坏了, 磨成非晶,衍射能力就没有了。磨得太狠的话,有些峰可能要消失了,而且相邻较近的衍射峰会由于宽化而相互叠加,最终会变成1个或几个"鼓包"。一般晶面间距大的峰受晶粒细化的影响会明显一些,因为d值大的晶面容易被破坏
衍射强度变弱本质的原因是由于晶体颗粒变小,还是样品颗粒变小?
强度除了和晶粒度有关外,还和晶粒的表面状态有关。一般颗粒越细,其表面积越大,表面层结构的缺陷总是比较严重的。结构缺陷将导致衍射强度降低和衍射峰宽化。XRD研究的应该是晶粒、晶体的问题,与晶体结构关联的问题,不是样品颗粒的大小问题,谢乐公式算的应该也是晶粒的大小。样品颗粒的大小要用别的方法测定,例如光散射、X射线散射、电镜等。
细针状微晶粉末样品做XRD重复性很差。(制作粉末衍射样品片)怎么可以避免择优取向?
择优取向是很难避免的,只能尽力减少他的影响。首先,你要讲样品磨得尽量细(但要适度,要注意样品的晶体结构不要因研磨过度而受到损坏);不要在光滑的玻璃板上大力压紧(压样时可以在玻璃板上衬一张粗糙的纸张),样品成形尽可能松一些;制样过程中也可以掺一些玻璃粉,或加一些胶钝化一下样品的棱角。当然还有其他一些方法,你可以上http://www.msal.net查找一下有关这方面的资料。
采用X射线进行晶体衍射分析,利用照相法记录衍射花样,1、当多晶体晶粒细化时,衍射花样将如何变化?
2、当多晶试样中存在宏观应力时,衍射花样的变化情况是怎样?
不管是粉末试样还是(多)晶体试样,粉末颗粒或晶粒太粗,参加衍射的晶粒少,会使衍射线条起麻,但粉末颗粒或晶粒过细时,会使衍射线条变宽,这些都不利于分析工作。存在宏观内应力的效应是使得衍射环或衍射峰的位置改变,导致底片上的衍射线条变宽,不利于分析工作。
XRD峰整体向右偏移是什么原因造成的?
可能是离子半径小的元素取代了离子半径大的元素。
也可能是你制样时,样品表面高出了样品座平面,或者仪器的零点不准造成的,建议你最好用标样来修正你的数据。
把样品靠后放置,使样品偏离测角仪中轴大概有1mm,请问衍射峰会怎么变化?
峰位移向低角度。样品表面偏离测角仪转轴0.1mm,衍射角的测量将产生约0.05?(2θ)的误差(对Cu靶,在2θ20?附近的位置)
影响仪器测量结果的分辨率仅仅取决于θ 吗?
影响仪器测量结果的分辨率的因素是多方面的:测角仪的半径;X射线源的焦斑尺寸;光学系统的各种狭缝的尺寸;仪器调整情况(2:1关系);采数步宽;样品定位情况等。
进行衍射分析时如何选择靶(X光管)?现有Cr的多晶试样,我只知道衍射分析时选Cr靶最好,但不知道为什么。
对于所有的元素,在高速电子的轰击下都会产生X射线还可能产生其特征X射线。元素受较高能量的X射线的照射时也能够激发其特征射线,称为二次X射线或荧光X射线,同时表现出对入射X射线有强烈的吸收衰减作用。
对于一定波长,随着元素周期表中的原子序数的增加其衰减系数也增加,但到某一原子序数时,突然降低;对于一定元素随着波长的增加质量衰减系数也会增加,但到某界限时质量衰减系数突然降低,此种情况可以出现数次。各元素的质量衰减系数突变时的波长值称为该元素的吸收边或吸收限。利用元素吸收性质的这个突变性质,我们能够为每一种X射线靶选择到一种物质做成"滤波片",它仅对该靶材产生的Kβ线强烈吸收而对其Kα波长只有部分的吸收,从而可以获得基本上由Kα波长产生的衍射图。原子序数比某靶材小1的物质正是这种靶的Kβ滤片。
但是,Kβ滤片不能去除样品产生的荧光X射线对衍射图的影响。荧光X射线的强度将叠加在衍射图的背景上,造成很高的背景,不利于衍射图的分析。因此,对于X射线衍射仪来讲,如果设备没有配置弯晶石墨单色器仅使用Kβ滤片,选波长(或者说选靶)主要考虑的就是样品中的主要组成元素不会受激发而产生强烈的荧光X射线。如果分析样品中的元素的原子序数比靶的元素的原子序数小1至4,就会出现强的荧光散射。例如使用Fe靶分析主要成分元素为Fe、Co、Ni的样品是合适的,而不适合分析含有MnCr V Ti的物质;Cu靶不适合于分析有Cr,Mn,Fe,Co,Ni这些元素的物质。所以,对于Cr是主要组成元素的样品,只能选择Cr靶X射线管。
石墨单色器不仅能够去除入射光束中的Kβ产生的衍射线,同时可以避免样品的荧光射线以及样品对"白光"产生的衍射叠加在衍射图的背景上,从而可以得到严格单色的Kα波长产生的衍射图。因此,在配置有弯晶石墨单色器的衍射仪上工作时,可以不用考虑样品产生的荧光X射线的干扰,Cu靶X射线管能够通用于各种样品,包括主要组成为Cr,Mn,Fe,Co,Ni等元素的样品。
但是具有波长大于CuKα波长的靶(如Cr、Fe、Co等靶)对于小角X射线衍射的研究或晶面间距的精确测定还是有价值的。因为波长增加能够减少衍射峰的重叠;使所有的衍射峰移向较高的角度。
(用X射线衍射仪)做X射线衍射时,一些(仪器)参数对谱线有什么影响?
如何选择仪器参数主要取决于做X射线衍射的目的。比如:扫描速度,如果是一般鉴定就可以取快一些(4-8度/分),如果是精修晶胞参数就要扫慢一些;狭缝条件,狭缝越小分辨率越高,但强度就会减小,不宜快扫,这又要增加时间。还要根据的样品的衍射能力、结晶度等因素来定。如果是步进扫描:测定晶胞参数可以取每个步进度1-30秒,如果做结构精测30秒到数分钟不等(对于普通功率的衍射仪,40kV、40mA)。
衍射峰左右不对称是何原因?
衍射仪获得的衍射峰形(精确地说是衍射线的剖面,diffraction lineprofile)是不对称的,尤其是在低角度区(2θ < 30°)表现更为明显。
峰型不对称是由多方面的因素造成的,主要是衍射仪光路的几何因素、仪器的调整状况以及样品的吸收性质等。
高精度测角仪是怎么实现θ/2θ倍角转动的?这种装置能够保证严格的倍角同步吗?
θ/2θ是两个同轴的园,θ是带动样品转动的园,而2θ是探测器转动的园,这样设计的目的是为了保证样品在转动中的衍射焦点始终在探测器转动的大园上。现代的衍射仪用2个步进电机分别独立控制θ和2θ园的转动,控制电路能够保证两个园按1:2转速比转动,保证两个园的转动严格倍角同步。
磁性材料比如NdFeB或者NdFeN的粉末,是不是会因为磁性的存在会产生择优取向?
磁性材料肯定是最具择优取向的,否则就没有磁性了,制样时应当磨成粉末,可以抑制这种取向趋势。"择优取向"会使很多本来有的衍射峰出不来。
为什么有的XRDdata中,有(200)(400)面,而没有最基本的(100)面数据?或者有(220)而没有(110)?
粉晶衍射不一定能出现所有的面网,很多物质的粉晶衍射都不一定出现(100)(110),这与结构有关。
晶体衍射有个叫"消光"的现象,晶体的"消光规律"决定于它的结构的对称性,不同的空间群其"消光规律"不同。
如果应该出现的衍射而没有出现,那就是样品的择优取向引起的。
再者(100)面的角度比较低,有时是没有扫到或淹没在低角度的背景中了。
[仪器技术]
介绍射线用晶体CdTe
CdTe是称为碲化镉的半导体化合物的一种。其具有对放射线有良好的吸收效率,由于能直接把光信号转换成电信号。被利用在作为放射线探出器的一种新型好材料。
至今为止,在探测放射线的元器件里有各种各样的探测材料,代表的有Lithium driftsilicone和锗。但是由于它们对放射线的感度不高,而且需要是使用液化氮气进行低之-196C的冷却。而且需要借助光敏管来转换光电信号。所以它们不易做到小型化。CdTe具有高感度,可以在常温的状态下发挥比以往产品更加具有优越的特性。对能量100keV放射线的只需要2mm厚的CdTe来制作探测器,感度是silicone的10倍。而且不需要冷却设备。CdTe可以用在射线的测量(诊断上)和低能射线的测定。使用CdTe探头的各类器械都能做到轻便,小型具有更高的分辨率的探测装置。
现在已经开发的产品有小型伽马照相机和各种X射线、γ射线探测器。在研究开发CT、PET的探测器的开发中已经走向成熟化。
常用公式-- 晶面夹角的计算公式 |
设晶面(h1k1l1)和晶面(h2k2l2)的面间距分别为d1、d2。则二晶面的夹角φ以下列公式计算(V为单胞体积)。 立方晶系:
|
立方晶系:
正方晶系:
六方晶系:
正交晶系:
单斜晶系:
三斜晶系: