原理
凡盐析所获得的粗制蛋白质(盐析得到的IgG)中均含有硫酸铵等盐类,这类将影响以后的纯化,所以纯化前均应除去,此过程称为“脱盐”(desalthing)。脱盐常用透析法和凝胶过滤法,这两种方法各有利弊。前者的优点是透析后析品终体积较小,但所需时间较长,且盐不易除尽;凝胶过滤法则能将盐除尽,所需时间也短,但其凝胶过滤后样品体积较大。所以,要根据具体情况选择使用。凝胶达滤后样品体积不会太增加,所以选用凝胶过滤法。
凝胶过滤(gel filtration),又称为凝胶层析(gelchromatography)、分子筛过滤(molecularsieve filtration)、凝胶渗透层析(gel osmoticchromatography)等。它是20世纪60年代发展起来的一种层析技术。其基本原理是利用被分离物质分子大小不同及固定相(凝胶)具有分子筛的特点,将被分离物质各成分按分子大小分开,达到分离的方法。
凝胶是由胶体粒子构成的立体网状结构。网眼里吸满水后凝胶膨胀呈柔软而富于弹性的半固体状态。人工合成的凝胶网眼较均匀地分布在凝胶颗粒上有如筛眼,小于筛眼的物质分子均可通过,大于筛眼的物质分子则不能,故称为“分子筛”。凝胶之所以能将不同分子的物质分开是因为当被分离物质的各成分通过凝胶时,小于筛眼的分子将完全渗入凝胶网眼,并随着流动相的移动沿凝胶网眼孔道移动,从一个颗粒的网眼流出,又进入另一颗粒的网眼,如此连续下去,直到流过整个凝胶柱为止,因而流程长、阻力大、流速慢;大于筛眼的分子则完全被筛眼排阻而不能进入凝胶网眼,只能随流动相沿凝胶颗粒的间隙流动,其流程短、阻力小、流速快,比小分子先流出层析柱;小分子最后流出。分子大小介于完全排阻不能进入或完全渗入凝胶筛眼之间的物质分子,则居中流出。这样被分离物质即被按分子的大小分开。
用于凝胶层析的凝胶均为人工合成的产品,主要有交联葡聚糖(商品名为Sephadex)、琼脂糖(商品名为Sepharose)、聚丙烯酰胺凝胶(商品名为Bio–gel)及具有一定网眼的细玻璃珠等和这些凝胶的衍生物。
下面主要介绍葡聚糖凝胶,这是由葡萄糖的多聚物与1–氯–2、3–环氧丙烷交连而成。环氧丙烷引入丙三醇基将链状的多聚葡萄糖单位交联起来,凝胶网眼的大小由多聚葡萄糖的分子和环氧丙烷的比例(交联度)来控制。
葡聚糖具有较强的亲水性,在水和电解质溶液中膨胀成为柔软而富于弹性的凝胶,其吸水能力与葡聚糖凝胶的交联度有密切关系。交联度大的,孔径小,吸水少,膨胀的程度小;交联度小的,孔径大,吸水多,膨胀的程度大。因此,葡聚糖凝胶孔径的大小可以其吸水量的大小来表示,常以G–10至G–200号码标记。G后面的数字是其吸水量(毫升水/克干胶)乘以10所得的值。如G–25即表示吸水量为2.5ml/2干胶。市售有G–10、G–5、G–50、G–75、G–100、G–150、G–200等型号。G–75以上的胶因吸水量大,膨胀后形态柔软易变,统称为软胶。G–75以下的称为硬胶。
葡聚糖凝胶可分离的分子大小从几百到数十万。可根据被分离物质的分子大小及目的选择使用。一般说SephadexG–l0~15通常用于分离肽及“脱盐”。Sephadex G–75~200用以分离各类蛋白质。
凝胶层析是一种物理分离法。葡聚糖凝胶基本上不带电荷呈多惰性,不与被分离物质发生反应,所以分离的效果较好。然而由于它是葡萄糖的聚合物,因而仍有少量活性羟基,能吸附少量蛋白质等被分离的物质。为了克服这个缺点,一般使用含有离子强度达0.08的NaCl等中性盐缓冲液作洗脱液。
操作方法
(1)取层析柱1支(1.5cm×20cm),垂直固定在支架上,关闭下端出口。将已经溶胀好的SephadexG-25中的水倾倒出去,加入2倍体积的0.0175mol/L,pH6.7磷酸盐缓冲液,并搅拌成悬浮液,然后灌注入柱,打开柱的下端出口,继续加入搅匀的SephadexG-25,使凝胶自然沉降高度到17cm左右,关闭出口。待凝胶柱形成后,在洗脱瓶中加入0.0175mol/L,pH6.7磷酸盐缓冲液以3倍柱体积的磷酸盐缓冲流过凝胶柱,以平衡凝胶。
(2)凝胶平衡后,用皮头滴管除去凝胶柱面的溶液,将盐析所得全部IgG样品加到凝胶柱表面,打开柱下口,控制流速让IgG样品溶液慢慢浸入凝胶内。凝胶柱面上加一层的0.0175mol/L,pH6.7磷酸盐缓冲液,并用此缓冲液洗脱,控制流速为0.5ml/min左右。用试管收集洗脱液,每管10滴。
(3)在开始收集洗脱液的同时检查蛋白质是否已开始流出。为此,由每支收集管中取出1滴溶液置于黑色比色磁盘中,加入1滴20%磺基水杨酸,若呈现白色絮状沉淀即证明已有蛋白质出现,直到检查不出白色沉淀时,停止收集洗脱液。
(4)由经检查含有蛋白质的每管中,取1滴溶液,放置在白色比色盘孔中,加入1滴奈氏试剂,若呈现棕黄色沉淀说明它含有硫酸铵。合并检查后不含硫酸铵的各管收集液,即为“脱盐”后的IgG。
注意:在检测时,用皮头滴管吸取管中溶液后应及时洗净,再吸取下一管,以免造成相互污染假象。
试剂和器材
(1)Sephadex G-25。
(2)0.0175mol/L,pH6.7磷酸盐缓冲液。
(3)奈氏(Nessler)试剂:于500ml锥形瓶内加入碘化钾150g,碘110g,汞150g及蒸馏水100ml。用力振荡7~15min,至碘的棕色开始转变时,混合液温度升高,将此瓶浸于冷水内继续振荡,直到棕色的碘转变为带绿色的碘化钾汞液为止。将上清液倾入2000ml量筒内,加蒸馏水至2000ml,混匀备用。
(4)20%(W/V)磺基水杨酸溶液。
(5)1.5cm×20cm层析柱。
(6)黑、白比色磁盘。
附:凝胶处理的基本操作
(1)凝胶溶胀(水化)商品葡聚糖凝胶和聚丙烯酰胺凝胶均为干燥颗粒。使用前必须水化溶胀。商品琼脂糖凝胶呈悬浮胶体可直接用,玻璃球不用溶胀。
凝胶溶胀有两种方法:一种是将所需葡聚糖凝胶浸入蒸馏水中于室温下溶胀;另一种是置于沸水浴中溶胀。各种葡聚糖在上述溶胀方法中所需的时间见下表:
型号溶胀时间(小时)20~25℃ 分离范围(蛋白质,Mr)
G-103 至700
G-153 至1500
G-253 1000~1500
G-503 1500~3000
G-7524 3000~7000
必须浸泡足够的时间以使凝胶充分溶胀。两种方法中,沸水浴溶胀不但节省时间,还可以杀灭凝胶中污染的细菌并排出网眼中气体。
凝胶溶胀后,需用蒸馏水洗涤几次,每次应将沉降缓慢的细小颗粒随水倾倒出去,以免在装柱后产生阻塞现象,降低流速。洗后将凝胶浸泡在洗脱液中待用。
一支层析柱中应该装入的干胶量可以用下法推算:称取1g所需型号的葡聚糖干胶,放在5ml量筒中,用室温溶胀的方法充分溶胀,观察溶胀后凝胶的体积。然后在层析柱中加水到所需柱床高度,将水倒出,量取柱床体积。根据1g干胶溶胀后的体积和所需柱床体积,即可推算出干胶的需要量。
(2)装柱将层析剂装入柱中进行层析的方法称柱层析法。作层析用的柱子称层析柱。层析柱有玻璃和透明塑料的两种。柱子的一端为进口,另一端为出口,出口端底部有烧结玻璃砂板或尼龙布,能阻止层析剂流出,溶剂则可流过。如果没有市售的层析柱,可以选用粗细均匀,长短合适的玻璃管,在两端塞上合适的胶塞,胶塞中插人细玻璃管,在一端放一层尼龙布为出口,即可作为层析柱使用。层析柱的长短粗细根据实验的目的而定,一般说来,凝胶层析时柱越长,分离效果越好。但柱过长,层析时间长,样品易稀释造成扩散,反而影响分离效果。柱的内径不宜较细,直径1cm以下的柱易发生“管壁效应”,即柱中部分的物质组分移动较快,管壁周围的则移动较慢,造成分离混乱。当然柱的内径也不宜过粗。
凝胶层析中,在把小分子物质(mw&1500),如无机或其他物质与大分子物质(mw&20000)分离时,层析柱的体积一般约为样品的4–10倍,高度与直径的比例为5:1至15:1之间。这类柱也称为“脱盐”柱,常用网孔很小的凝胶如G–25。用以将生物大分子物质彼此分开的分级层析柱,体积应为样品体积的25~100倍。柱长度与直径的比例为20:1~100:1。
装柱的操作过程如下:将层析柱垂直固定在支架上,打开柱下口开关。将溶胀好的凝胶放在烧杯中,使凝胶表面上的水层与凝胶体积相等。用玻璃棒搅匀凝胶液,顺玻璃棒灌入柱内。此时柱下口一边排水,上口一边加入搅匀的凝胶,可见凝胶连续均匀地沉降,逐步形成凝胶柱。当到达所需凝胶柱高度时,立即关闭下口,待凝胶自然沉降形成凝胶柱床。凝胶柱床一般应离柱顶3~5cm,并覆盖一层溶液。
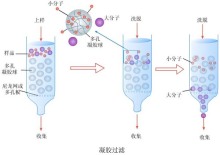
灌注凝胶时要求将均匀的凝胶一直加到所需柱床高度,不能时断时续,否则将出现分层或“纹路”等毛病。若中途出现这些现象,可以用玻璃棒将已形成的柱床逐步搅起,直至出毛病的部分再让凝胶重新沉降或继续加入搅匀的凝胶悬液。若在灌好胶后才发现“纹路”、分层等现象时,要重新装柱,以免影响层析效果。在做大型的凝胶柱时,灌注的凝胶是否均匀往往从表面上看不出来。所以使用前应该用一些带色的大分子物质如细胞色素C、血红蛋白或专用的蓝色葡聚糖–2000 (Blue dextran–2 000)通过凝胶柱,观察形成的色斑带是否整齐,若斑带歪斜,应该重新装柱,直至达到要求。
刚从冰箱中取出的凝胶液,不能马上用来灌柱,应平衡至室温后再用,不然装好的凝胶会产生大量的气泡影响层析。
在整个灌注凝胶的过程及使用中,凝胶柱面上一定要覆盖着一层溶液,以免进入空气。若进空气再加入溶液时,凝胶柱中易形成气泡。
(3)平衡装好的凝胶柱,使用前应该用相当于柱床体积两倍或更多的洗脱液流过凝胶柱,以压实凝胶,称为平衡。
(4)上样将样品加入到凝胶柱中,准备层析的过程称上样。上样时,应该注意上样量的多少、样品的黏稠度及离子强度。这三个因素会影响到以后层析的效果。一般来说“脱盐”层析时,上样体积可为柱体积的10%~25%;生物大分子的分级分离,约为柱体积的1%~5%。样品的黏稠度一般不宜大于洗脱液黏稠度的2倍以上,不然洗脱峰会变宽和歪斜。离子强度要达到0.08,以免产生特异性吸附。
上样操作:先打开层析柱的下口开关,放出凝胶柱面上的溶液,或用皮头吸管吸取,使液面与凝胶表面相平齐,但切忌液面低于凝胶表面。然后将样品加在凝胶表面,打开柱下口开关,控制流速,使样品慢慢渗入凝胶内。加样时注意勿将凝胶柱面冲起形成凹面,也不能沿管壁流下,以免样品沿柱壁与凝胶柱的间凝胶隙漏下。当慢慢渗入凝胶的样品液液面与凝胶柱面相平时,关闭下口,完成上样。然后在凝胶柱面上加一层(3~5cm)洗脱液,接上洗脱瓶准备洗脱。
(5)洗脱用规定的溶液流过样品,使分子量不同的样品逐步分开并先后由柱床流出的过程称为洗脱。所用溶液称洗脱液。洗脱液放在贮液瓶(洗脱瓶)中并与层析柱相通连。洗脱时只要打开层析柱下口开关,洗脱液即可流出。
洗脱过程中保持恒定的流速是柱层析获得良好分离效果的重要条件。因为凝胶层析的分离作用主要取决于分子扩散进入凝胶的机会,流速过快有些分子来不及在分子筛中分配而流出;过慢时已分离的分子会因扩散而混合。因而要保持适当的恒定流速。洗脱液的流速取决于它的静水压。静水压是指洗脱瓶中洗脱液的液面高出于层析柱出口产生的液体压力(或接触空气的两个液面间的高差),这个高度差越大,静水压越大,洗脱液的流速就越快。这个压力差可以调动,如提高洗脱瓶的位置或瓶中液面的增减;或者降低或提高层析柱出口的位置等,因此静水压又称操作压。
由于静水压越大,洗脱液的流速就越快。在固定洗脱瓶与出口位置的情况下,静水压会在洗脱过程中,随着洗脱瓶中溶液减少液面不断下降而减少,难以维持流速的恒定。为了解决这个问题,Marriotte首次将洗脱瓶加塞塞紧,塞中插入玻璃管,使玻管下口深入到洗脱液的底部,这样洗脱液的静水压便等于此玻管下口的液面高出层析柱出口液面的高度(因为当洗脱瓶液体流出时,瓶顶形成真空,空气只从玻璃管中进入,玻管下口即为接触空气点),因此只要溶液不低于玻璃管下口,玻璃管口以上溶液的增减将不影响静水压,从而自动保持了流速的恒定,此洗脱瓶称为恒压瓶,也叫马氏瓶。当然,当洗脱液减少至液面低于玻管的下口时,便会失去作用,但这时可用及时加入洗脱液来解决。
在凝胶层析中,不仅要保持静水压的恒定,还要保持适当的静水压。这是因为G–75以上的软胶胶体柔软易变形,对静水压仅有一定的承受能力,不同软胶的承受能力也不相同,静水压超过凝胶的承受能力时,最初流速会很快;其后随着静水压力过大将使软胶变形压紧,流速逐渐降低,最后甚至流不出。所以,保持恒定正确的静水压是凝胶层析时获得满意结果的必要条件。
脱盐一般使用的是G–25属于硬胶,硬胶不易变形,受静水压的影响较小,实验过程中的流速可用恒压瓶下口橡皮管上的螺旋夹控制在0.5ml/min左右,随着洗脱液的流出,样品逐步被分开。用试管收集洗脱液,按顺序每管收集2ml。
(6)检测在收集的同时,检查蛋白质是否流出。于每管中取出1滴放在黑色比色板孔中(按编号顺序),再分别加入1滴20%磺酰水扬酸,如出现白色沉淀即表示蛋白质已流出凝胶柱,如此一直检查到蛋白质全流出为止。与此同时,再从蛋白质的管中取出1滴于白色比板中,加入奈氏试剂1滴,如蛋白管中不出现棕色,即表示蛋白质中的硫酸铵已除去。合并纯化后的蛋白质管以待用。以光密度值为纵坐标,以收集管的顺序号为横坐标,在坐标纸上绘图。可获具有一条洗脱峰的曲线,称为洗脱曲线,用以表示被分离物质流出的状态。
(7)凝胶的洗涤及保存由于凝胶对被分离的物质基本无吸附作用,所以无需“再生”,只要用洗脱液冲洗后即可连续使用。但因多次使用凝胶颗粒将逐渐沉积压紧,流速将减慢,所以使用一段时间后应倒出来,清洗后重新装柱。
凝胶暂时不使用可浸泡在溶液中,存放在4℃冰箱中。若放在室温保存应加入0.02%叠氮钠(NaN3)或0.01%乙酸汞等防腐剂,以防发霉。用时以水洗去防腐剂即可使用。凝胶长期不用,可用水洗净,分次加入百分浓度递增的乙醇溶液,每次停留一段时间,使之平衡,再换一下浓度的乙醇,让凝胶逐步脱水,再用乙醚除乙醇,抽干即可,或将凝胶洗净后抽干,在表面皿上30℃逐步烘干。



