有些网友觉得pcr技术论文难写,可能是因为没有思路,所以小编为大家带来了相关的例文,希望能帮到大家!
PCR技术的研究与现状分析篇一
摘要:PCR技术是一种体外酶促合成、扩增特定DNA片段的方法。因其高强的特异性和灵敏度以及检测速度快、准确性好等优点,该技术已经广泛地应用于水产、微生物检测、临床微生物、基因表达、肿瘤免疫、微小残留病变的检测,DNA拷贝数的测量、基因组变异和多态性等许多方面。本文主要介绍4种PCR技术及其在各个领域的应用现状。 关键词:PCR技术;分类;研究现状;应用;
Research and Application Status of PCR Technology
Class 1 Bio-engineering 10 Student: Lao Yangyan Student ID: 1031250019 Tutor: Wei Dongmei Abstract: PCR is an in vitro enzymatic synthesis,amplification of specific DNA fragment. Because of its high-strength specificity and sensitivity,detection speed and good accuracy,it has been widely applied in many fields such as the aquatic,microbial detection,clinical
microbiology,gene expression,tumor immunology,detect ion of minimal residual lesion,measurement of DNA copy number,genome mutation,polymorphism and so on. This article summarize 4 kinds of PCR technology,with its application status is analysed.
Keywords: PCR technology;Category;Progress research;Application
引言
聚合酶链反应( PCR) 技术问世20多年以来, 已经在生物学研究领域内得到了巨大的发展并不断的完善, 不仅广泛应用在基础研究领域, 在疾病诊断等方面也有了越来越广泛深入的研究和应用。1985年,美国Karray等学者首创了PCR 技术,并且由美国Cetus 公司开发研制[1]。随着科学技术的发展和突破,PCR 技术已在多个领域得到广泛地应用,如微生物检测、兽医学、水产养殖等方面。由于该技术具有较强的灵敏度、准确度和特异性,又能快速进行检测,因而其应用领域也在不断延伸[2]。随着PCR 技术的不断发展,在常规PCR 技术的基础上又衍生出了许多技术,如多重PCR技术、实时荧光定量PCR技术、单分子PCR 技术、变性梯度凝胶电泳技术。目前应用最多的为荧光定量。本文主要介绍4种PCR技术及其在各个领域的应用现状。并对PCR的前景进行了展望,让其更好的服务于人们的生活。 1 PCR技术原理
PCR 技术是根据待扩增的已知DNA片段序列、人工合成与该DNA 2条链末端互补的2段寡核苷酸引物,在体外将待检DNA序列(模板)在酶促作用下进行扩增。PCR 的整个技术过程经若干个循环组成,一个循环包括连续的3个步骤:第1步是高温条件下的DNA模板变性,即模板DNA在93~94℃的条件下变性解链;第2步是退火,即人工合成的2个寡核苷酸引物与模板DNA链3’端经降温至55℃退火;第3步是延伸,即在4种dNTP底物同时存在的情况下,借助TaqDNA聚合酶的作用,引物链将沿着5’-3’方向延伸与模板互补
的新链[3]。经过这个循环后,合成了新链,可将其作为DNA模板继续反应,由此循环进行。循环进程中,扩增产物的量以指数方式增加,一般单一拷贝的基因循环25~30次,DNA可扩增l00 ~200万倍。PCR反应的步骤很简单,但是具体的操作是复杂的,如退火温度的确定、延伸时间的长短以及循环数等。因此,不同的反应体系应该确定适当的反应条件,以避免假阴性或假阳性等情况的产生。
2 PCR技术的分类
在传统PCR 技术的基础上,根据人们的需要以及各个领域的应用要求,又衍生出很多种类的PCR 技术。新技术在各领域广泛应用并逐渐改进,为进一步的研究提供了基础。
2.1 实时荧光定量PCR技术
1996 年,学者经过研究,在传统PCR 技术的基础上,首创了实时荧光定量PCR 技术,新技术已经应用至医学领域、分子生物学和其他基础研究领域。实时荧光定量PCR 技术基于传统技术的优势,还具有实时性、准确性、无污染,实现了自动化操作和多重反应,是PCR 技术研究史上从定性到定量的飞跃[4]。荧光定量PCR 技术最大的特点是能将荧光基团加入到PCR 反应体系中,借助于荧光信号,累积实时监测整个PCR进程,最后通过标准曲线对未知模板进行定量分析[5]。实时监测这一特点是常规PCR 技术所不具有的,因为其对扩增反应不能进行随时的检测。常规PCR 技术的扩增终产物需要在凝胶电泳等条件下才能进行,无法对起始模板进行准确的定量,而荧光定量PCR 技术的反应进程可以根据荧光信号的变化做出准确的判断[6]。一个PCR 循环反应结束之后,定量PCR 仪可以收集1个荧光强度信号,荧光信号强度的变化可以反映产物量的变化情况,这样就可以得到1条荧光扩增曲线[7]。荧光信号在指数扩增阶段,PCR 产物荧光信号的对数值与起始模板量之间存在线性对应关系,然后进行定量分析[8]。
2.2 多重PCR 技术
多重PCR(mutiplex PCR)技术是PCR 技术的一种,为同一管中加入多对特异性引物,与PCR 管内的多个模板反应,在一个PCR 管中同时检测多个目标DNA分子。多重PCR技术可以扩增一个物种的一个片段,也可以同时扩增多个物种的不同片段[9]。在同一反应体系中,多重PCR 技术进行多个位点的特异性扩增时,引物间的配对、引物间的竞争性扩增等会对扩增效果产生重要影响。一方面,如果能选择适宜的反应体系和反应条件,可极大地提高多重PCR 的扩增效果[10]。主要包括退火温度、退火及延伸时间、PCR 缓冲液成分、dNTP 的用量、引物及模板的量等。另一方面,DNA的抽提质量也影响多重PCR 扩增效率,如DNA抽提不干净或降解都将影响PCR 扩增效果[11]。
2.3 单分子PCR 技术(SM-PCR)
单分子PCR 技术是在传统PCR 技术的基础上发展的,基本循环过程相同,但在反应条件、模板数量、DNA聚合酶选择、引物设计方面具有不同点。该技术是以少量或单个DNA分子为模板进行的PCR[12]。单分子PCR 技术反应中,DNA模板浓度极低,这就要求模板有较高的质量。因为这是试验成败的决定性因素。在设计引物时,应该严格控制GC 的含量和Tm值,同时尽量避免引物间存在可配对序列。在反应混合物模板数极低的情况下,若引物之间存在少量配对序列,扩增时极易形成二聚体,使反应无法进行,得不到所需要的产物[13]。由于单分子PCR 技术反应的变性温度(96~98 ℃)大多比常规PCR 技术(94 ℃)略高,因而对DNA聚合酶热稳定性的要求也更加严格,需要有较好的热稳定性,以防止温度过高而使其失活。其变性时间(5~15 s)、退火时间及延伸时间也短于常规PCR技术。
2.4 变性梯度凝胶电泳PCR技术(Denaturing Gradient Gel Electrophoresis)
PCR-DGGE 技术是基于核酸序列的不同,将片段大小相同的DNA序列分开的一种技术方法[14]。在进行变性梯度凝胶电泳时,序列不同的DNA片段因为碱基组成和排列的差异,在聚丙烯酰胺凝胶中解链时需要不同的变性剂浓度,并会发生空间构型的变化,最终导致电泳迁移率的差异。将通过PCR扩增之后得到的等长双链DNA分子,在含梯度变性剂( 如尿素、甲酰胺) 的聚丙烯酰胺凝胶中进行电泳时,电泳迁移率的差异会使不同序列的DNA片段停留在凝胶的不同位置,从而形成相互分开的条带图谱[15]。从理论上讲, 只要选择的电泳条件足够精细, 就可以分开仅有1个碱基差异的DNA片段[ 16]。
3 PCR技术的应用
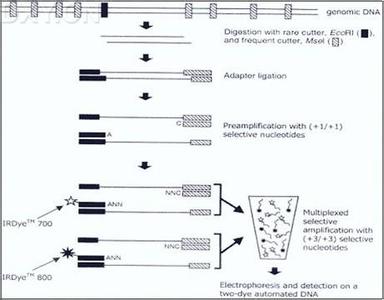
3.1 PCR技术在水产上的应用
基因表达是检测某个基因在不同发育期或不同组织中的表达量变化,或受到某种试验处理过程中的影响而出现表达量变化的情况。有学者应用real-time PCR 技术研究碳水化合物含量对翘嘴红鲴糖代谢酶G6Pase、GK 以及PEPCK表达量的影响[17],研究结果可为翘嘴红鲴饲料配方中的最合适糖含量提供理论依据[18]。孙淑娜等研究叶酸拮抗剂对斑马鱼心脏发育相关基因BMP2b及HAS2表达的影响,表明叶酸拮抗剂对早期胚胎的心脏发育影响较大,可导致斑马鱼心脏发育延迟及心脏形态异常,并下调斑马鱼心脏发育相关基因BMP2b 及HAS2 的表达,这可能是叶酸生物学活性受抑后导致心脏发育异常的机制之一。Sawyer et al以斑马鱼的未受精卵、胚胎、仔鱼和成鱼为研究材料,采用实时荧光定量PCR 技术,检测了P450aromA和P450aromB 在不同组织的表达量,表明在各组织中均有2种基因的表达,但表达量显著不同,呈现组织特异性。
3.2 PCR技术在食品微生物检测中的应用
实时荧光定量PCR技术,是指在PCR反应体系中加入荧光染料或荧光探针,利用荧光信号积累实时监测整个PCR进程,最后通过标准曲线对未知模板进行定量分析的方法。该技术实现了PCR 从定性到定量的飞跃,除了具有常规PCR的快速、灵敏、操作方便等特点外,还具有以下优点:全封闭反应,无需凝胶电泳等PCR后处理,减小对环境的污染;不使用有毒试剂,操作安全;定量准确;仪器在线实时监测,结果直观。
多重PCR技术是通过对普通PCR技术的改进而建立起来的一种技术,是将多条引物和多条模板混合在一个反应体系中分别特异扩增不同的目的条带,或者多条引物和单一的模板DNA混合在同一反应体系中扩增同一模板的不同片断,常用于对超长片断的扩增。与常规的PCR相比,还具有扩增效率高、产物特异性高和经济简便等特点,特别适合食品中多种致病菌的快速检测。
PCR-DGGE技术具有可检测不可培养类细菌、检测时间短、检测率高、重复性好等优点。近些年来,此技术在食品中检测微生物后应用也有报道。
3.3 PCR技术在临床微生物中的应用
许多传染病的特点是具有高度的变异率,这会严重影响对致病菌的评估,分子信标可以用于病原菌的定量。它的优点是: 一个没有荧光的淬灭物可以用于4个不同的荧光染料, 这样就可以同时检测和定量4个样本。在许多应用领域需要同时对多个核酸进行定量, 这将大大降低检测的花费和所需的时间。这个方法的另一个好处在于加样的误差被最小化了, 而且所有核酸都是在相同的条件下同时扩增, 这样它所获得的数据就更加精确可靠。当然, 多通道检测比单通道检测更为复杂也需要解决可能出现的更多问题。广东省花都市人民医院、中山医科大学基因诊断中心采用荧光定量PCR 检测技术对淋球菌、沙眼衣原体、解脲支原体、人类乳头瘤病毒、单纯疱疹病毒等5个项目进行了定量测定, 结果表明定量PCR方法可靠性和重复性好,且操作简便、快速,结果判断客观。除此以外,目前已有用此方法对人类免疫缺陷病毒,肝炎病毒,结核杆菌,巨细胞病毒,EB病毒,流感病毒A、B等病原体进行检测的报道。
3.4 PCR技术在畜牧兽医领域的应用
家育DNA多态性研究是近年来兴起的分子水平遗传标记选择技术, 对于开展品种问遗传差异程度、种群间亲缘关系、个体问遗传相似性, 亲子鉴定等方面的工作, 对于开展数量性状的间接选择, 早期选择, 提高数量性状选择效率等研究均具有重要的意义和潜在的应用价值。PCR技术的出现大大推动了上述研究的进程。以PCR技术为基础的DNA多态性分析和检测技术, 如PCR-RFLP(限制性长度多态性) , PCR-SSCP (多聚酶链反应产物的单链构象多态性) 和AFLP(扩增片断长度多态性)等, 已经在猪DNA多态性研究中得到了应用, 并且有可能迅速推广到育种实践中去。
3.5 PCR技术在转基因食品检测中的应用
转基因食品,是利用基因工程技术将有利的基因转移到微生物,,植物或动物细胞内,使他们获得有利的特性,再由这些转基因物种生产或处理获得的食品及添加剂。由此可增加食品的种类,提高产量,改进营养成分的构成,延长货架期等,目前,转基因作物及其产品的安全性问题越来越引起消费者的重视[25]。为了保护人类的健康,世界卫生组织在20世纪90年代提出了对转基因食品进行安全性评价的要求,对食品中的GMOs存在与否进行标示。因此,对食品中的GMOs 进行监测,建立安全性检验的技术与方法,成为管理和审批工作的重要基础。在众多的监测方法中,PCR 是最常用以及最适用的监测转基因食品的方法。PCR 方法中,常用的几个特定序列分为三类:
1、调控序列:他们调节转入植物的基因的表达。由于大多数转基因食品的基因都含有CaMV 35S 启动子和NOS 终止子,因此,在检测食品中是否含有GMOs 时,可以直接检测CaMV 35S 启动子和/或NOS 终止子的存在与否,如果存在,即可说明其中存在GMOs。
2、标记序列,用于帮助在遗传转化中筛选和鉴定转化的细胞,组织和再生植株,一般是抗生素抗性基因。
3、目的基因,即转入的基因,如抗虫、抗除草剂基因。用PCR 方法鉴定植株中检出的目的基因,标记基因,报告基因,35S,NOS 等外源基因片段,为转基因食品的筛选鉴定及为安全性评价提供敏感直接的数据,成为转基因食品安全性检验中一项重要的技术手段。 4 PCR技术的应用前景展望
传统PCR技术以及衍生出来的新型PCR技术自面世以来,已被广泛应用到生命科学的各个领域。随着技术方法的不断改进与完善,荧光定量PCR技术将会逐渐完善并广泛应用;多重PCR技术在食品病原微生物、非致病微生物及环境微生物检测中将具有重要作用。未来的研究主要集中在去除食品抑制因子干扰、改进样品前处理技术等方面。
随着研究的不断深入发展与完善,许多相关的新类型及改良的PCR技术将会不断涌现,这些派生出的新类型和新方法必将在未来食品检测中有非常好的应用前景。
参考文献:
[1] 常世敏, PCR在食品微生物检测中的应用[J]. 邯郸农业高等专科学校学报, 2004, 21(4): 23-25.
[2] 唐永凯, 俞菊华, 徐跑, 等. 实时荧光定量PCR技术及其在水产上的应用[J].中国农学通报, 2010(21):422-426.
[3] 谢海燕.黑线仓鼠LHR 部分序列克隆及组织器官的表达差异[D].曲阜:曲阜师范大学,2011.
[4] KUBISTA M, ANDRADE J M,BENGTSSO N M, et a1.The real-time polymerase chain reaction[J].
MoLecular Aspects of Medicine, 2006, 27(2-3): 95-125.
[5] AGINDOTAN B O, SHIEL P J, BERGER P H. Simultaneous detection of potato viruses, PLRV, PVA, PVX
and PVY from dormant potato tubers by TaqMan real-time RT-PCR[J]. J Virol Methods, 2007, 142(1-2): l-9.
[6] 薛霜, 独军政, 高闪电, 等.实时荧光定量PCR技术研究进展及其在兽医学中的应用[J]. 中国农学通报,2010(7): 11-15.
[7] SCHUBERT J, FOMITCHEVAV, SZTANGRET-WISNIEWSKAJ. Differentiation of Potato virus Y strains
using improve dsets of diagnostic-PCR-primers [J]. J Virol Methods, 2007, 140(1-2): 66-74.
[8] 袁继红. 实时荧光定量PCR技术的实验研究[J]. 现代农业科技, 2010(13): 20-22.
[9] 朱善元. 生物检测技术PCR及其在兽医微生物检测中的应用[J]. 黑龙江畜牧兽医, 1999(11): 21-22.
[10] 银 花, 胡晓湘, 李 宁, 等. 影响多重PCR扩增效果的因素[J]. 遗传, 2003, 25(1): 65-68.
[11] 陈 诺, 唐善虎,岑璐伽,等.多重PCR技术在食品微生物检测中的应用进展[J].生物技术, 2010, 37(10):72-75.
[12] 刘连生. 常规PCR技术与单分子PCR技术[J]. 医学信息, 2010, 23(11): 4379-4380.
[13] 顾超颖. 汗孔角化病的临床分析, SSH1、ARPC3 基因突变检测和表达谱分析[D]. 上海: 复旦大学,
2008.
[14] Muyzer G, DeWaal E C, Uitterlinden AG. Profiling if complex microbial population by denaturing gradient
gel electrophoresis analysis of polymerase chain react ion-amplified genes encoding for 16S rDNA[J] . Applied and Environmental Microbiology, 1993, 59( 3) : 695- 700.
[15] Muyzer G, SmallAK. Application of denaturing gradient gel electrophoresis( DGGE ) and temperature
gradient gel electrophoresis ( TGGE) in microbial ecology [J] . Antonie Van Leeuwenhoek , 1998, 73: 127-141.
[16] 吴高锋, 李文刚, 高卫科, 等. PCR-DGGE的原理及在动物肠道菌群分析中的应用[ J] . 中国畜牧兽
医, 2008, 35( 6) : 37-39.
[17] 唐永凯, 俞菊华, 刘波, 等. 翘嘴红鲌肝脏G6Pase催化亚基的克隆以及摄食和饲料中碳水化合物对其
表达的影响[J]. 水产学报, 2007, 31(1): 45-53.
[18] 刘波, 谢骏, 苏永腾, 等. 高碳水化合物日粮对翘嘴红鲌生长、GK 及GK mRNA表达的影响[J]. 水
生生物学报, 2008, 32(1): 47-53.
pcr技术论文篇二
摘要:PCR技术又称无细胞分子克隆系统或特异性DNA序列体外引物定向酶促扩增法,是体外酶促合成特异DNA片段的一种方法,由高温变性、低温退火及适温延伸等几步反应组成一个周期,循环进行,使目的DNA得以迅速扩增,具有特异性强、灵敏度高、操作简便、省时等特点,是基因扩增技术的一次重大革新。PCR技术可将极微量的靶DNA特异地扩增上百万倍,从而大大提高对DNA分子的分析和检测能力,能检测单分子DNA或对每10万个细胞中仅含1个靶DNA分子的样品,因而此方法立即在分子生物学、微生物学、医学及遗传学等多领域广泛应用和迅速发展。 关键词:PCR原理、PCR过程及应用、关于PCR技术要点
1、PCR技术的基本原理
DNA的半保留复制是生物进化和传代的重要途径。双链DNA在多种酶的作用下可以变性解链成单链,在DNA聚合酶与启动子的参与下,根据碱基互补配对原则复制成同样的两分子挎贝。在实验中发现,DNA在高温时也可以发生变性解链,当温度降低后又可以复性成为双链。因此,通过温度变化控制DNA的变性和复性,并设计引物做启动子,加入DNA聚合酶、dNTP就可以完成特定基因的体外复制。
PCR的过程类似于DNA的天然复制过程,其特异性依赖于与靶序列两端互补的寡核苷酸引物。PCR由变性--退火(复性)--延伸三个基本反应步骤构成:①模板DNA的变性:模板DNA经加热至93℃左右维持一定时间,使含有所需扩增分析序列的靶DNA双链经热变性处理解开为两个寡聚核苷酸单链,然后加入一对根据已知DNA序列由人工合成的与所扩增的DNA两端邻近序列互补的寡聚核苷酸片段作为引物,即左右引物。此引物范围就在包括所欲扩增的DNA片段,一般需20-30个碱基对,过少则难保持与DNA单链的结合;②模板DNA与引物的退火(复性):模板DNA经加热变性成单链后,温度降至55℃左右,引物与模板DNA单链的互补序列配对结合;③引物的延伸:DNA模板--引物结合物在TaqDNA聚合酶的作用下,于72℃左右,以4种三磷酸脱氧核苷(dNTP)为反应原料,靶序列为模板,按碱基配对与半保留复制原理,按5'到3'方向将引物延伸、自动合成新的DNA链、使DNA重新复制成双链。重复循环变性--退火--延伸三过程,就可获得更多的“半保留复制链”, 每次循环延伸的模板又增加1倍,亦即扩增DNA产物增加1倍,经反复循环,使靶DNA片段指数性扩增。每完成一个循环需2~4分钟,2~3小时就能将待扩目的基因扩增放大几百万倍。
2、PCR技术的反应体系与反应条件
(一)标准的PCR反应体系:
10×扩增缓冲液 10ul
4种dNTP混合物 各200umol/L
引物 各10~100pmol
模板DNA 0.1~2ug
Taq DNA聚合酶 2.5u
2+ Mg 1.5mmol/L
加双或三蒸水至 100ul
(二)PCR反应五要素: 参加PCR反应的物质主要有五种即引物、酶、dNTP、模板和Mg
(1)引物: 引物是PCR特异性反应的关键,PCR 产物的特异性取决于引物与模板DNA互补的程度。理论上,只要知道任何一段模板DNA序列,就能按其设计互补的寡核苷酸链做引物,利用PCR就可将模板DNA在体外大量扩增。
设计引物应遵循以下原则:
①引物长度: 15-30bp,常用为20bp左右。
②引物扩增跨度: 以200-500bp为宜,特定条件下可扩增长至10kb的片段。
③引物碱基:G+C含量以40-60%为宜,G+C太少扩增效果不佳,G+C过多易出现非特异条带。ATGC最好随机分布,避免5个以上的嘌呤或嘧啶核苷酸的成串排列。
④避免引物内部出现二级结构,避免两条引物间互补,特别是3’端的互补,否则会形成引物二聚体,产生非特异的扩增条带。
⑤引物3’端的碱基,特别是最末及倒数第二个碱基,应严格要求配对,以避免因末端碱基不配对而导致PCR失败。
⑥引物中有或能加上合适的酶切位点,被扩增的靶序列最好有适宜的酶切位点,这对酶切分析或分子 克隆很有好处。
⑦引物的特异性:引物应与核酸序列数据库的其它序列无明显同源性。
⑧引物量: 每条引物的浓度0.1~1umol或10~100pmol,以最低引物量产生所需要的结果为好,引物浓度偏高会引起错配和非特异性扩增,且可增加引物之间形成二聚体的机会。
(2)酶及其浓度。目前有两种Taq DNA聚合酶供应, 一种是从栖热水生杆菌中提纯的天然酶,另一种为大肠菌合成的基因工程酶。催化一典型的PCR反应约需酶量2。5U(指总反应体积为100ul时),浓度过高可引起非特异性扩增,浓度过低则合成产物量减少。
(3)dNTP的质量与浓度。dNTP的质量与浓度和PCR扩增效率有密切关系,dNTP粉呈颗粒状,如保存不当易变性失去生物学活性。dNTP溶液呈酸性,使用时应配成高浓度后,以1M NaOH或1M Tris。HCL的缓冲液将其PH调节到7.0~7.5,小量分装, -20℃冰冻保存。多次冻融会使dNTP降解。在PCR反应中,dNTP应为50~200umol/L,尤其是注意4种dNTP的浓度要相等( 等摩尔配制),如其中任何一种浓度不同于其它几种时(偏高或偏低),就会引起错配。浓度过低又会降低PCR产物的产量。dNTP能与Mg结合,使游离的Mg浓度降低。
(4)模板(靶基因)核酸。模板核酸的量与纯化程度,是PCR成败与否的关键环节之一,传统的DNA纯化方法通常采用SDS和蛋白酶K来消化处理标本。SDS的主要功能是: 溶解细胞膜上的脂类与蛋白质,因而溶解膜蛋白而破坏细胞膜,并解离细胞中的核蛋白,SDS 还能与蛋白质结合而沉淀;蛋白酶K能水解消化蛋白质,特别是与DNA结合的组蛋白,再用有机溶剂酚与氯仿抽提掉蛋白质和其它细胞组份,用乙醇或异丙醇沉淀核酸。提取的核酸即可作为模板用于PCR反应。一般临床检测标本,可采用快速简便的方法溶解细胞,裂解病原体,2+2+2+消化除去染色体的蛋白质使靶基因游离,直接用于PCR扩增。RNA模板提取一般采用异硫氰酸胍或蛋白酶K法,要防止RNase降解RNA。
(5)Mg浓度。Mg对PCR扩增的特异性和产量有显著的影响,在一般的PCR反应中,各种dNTP浓度为200umol/L时,Mg浓度为1.5~2.0mmol/L为宜。Mg浓度过高,反应特异性降低,出现非特异扩增,浓度过低会降低Taq DNA聚合酶的活性,使反应产物减少。
(三)PCR反应条件的选择:
PCR反应条件为温度、时间和循环次数。
温度与时间的设置:基于PCR原理三步骤而设置变性-退火-延伸三个温度点。在标准反应中采用三温度点法,双链DNA在90~95℃变性,再迅速冷却至40 ~60℃,引物退火并结合到靶序列上,然后快速升温至70~75℃,在Taq DNA 聚合酶的作用下,使引物链沿模板延伸。对于较短靶基因(长度为100~300bp时)可采用二温度点法, 除变性温度外、退火与延伸温度可合二为一,一般采用94℃变性,65℃左右退火与延伸(此温度Taq DNA酶仍有较高的催化活性)。
①变性温度与时间:变性温度低,解链不完全是导致PCR失败的最主要原因。一般情况下,93℃~94℃lmin足以使模板DNA变性,若低于93℃则需延长时间,但温度不能过高,因为高温环境对酶的活性有影响。此步若不能使靶基因模板或PCR产物完全变性,就会导致PCR失败。
②退火(复性)温度与时间:退火温度是影响PCR特异性的较重要因素。变性后温度快速冷却至40℃~60℃,可使引物和模板发生结合。由于模板DNA 比引物复杂得多,引物和模板之间的碰撞结合机会远远高于模板互补链之间的碰撞。退火温度与时间,取决于引物的长度、碱基组成及其浓度,还有靶基序列的长度。对于20个核苷酸,G+C含量约50%的引物,55℃为选择最适退火温度的起点较为理想。引物的复性温度可通过以下公式帮助选择合适的温度:
Tm值(解链温度)=4(G+C)+2(A+T)
复性温度=Tm值-(5~10℃)
在Tm值允许范围内, 选择较高的复性温度可大大减少引物和模板间的非特异性结合,提高PCR反应的特异性。复性时间一般为30~60sec,足以使引物与模板之间完全结合。
③延伸温度与时间:Taq DNA聚合酶的生物学活性:
70~80℃ 150核苷酸/S/酶分子
70℃ 60核苷酸/S/酶分子
55℃ 24核苷酸/S/酶分子
高于90℃时, DNA合成几乎不能进行。
PCR反应的延伸温度一般选择在70~75℃之间,常用温度为72℃,过高的延伸温度不利于引物和模板的结合。PCR延伸反应的时间,可根据待扩增片段的长度而定,一般1Kb以内的DNA片段,延伸时间1min是足够的3~4kb的靶序列需3~4min;扩增10Kb需延伸至15min。延伸进间过长会导致非特异性扩增带的出现。对低浓度模板的扩增,延伸时间要稍长些。 2+2+2+2+循环次数的设置:循环次数决定PCR扩增程度。PCR循环次数主要取决于模板DNA的浓度。一般的循环次数选在30~40次之间,循环次数越多,非特异性产物的量亦随之增多。
3、PCR反应的基本步骤
标准的PCR过程分为三步:
1。DNA变性(90℃-96℃):双链DNA模板在热作用下,氢键断裂,形成单链DNA。
2。退火(复性)(25℃-65℃):系统温度降低,引物与DNA模板结合,形成局部双链。
3。延伸(68℃-75℃):在Taq酶(在72℃左右最佳的活性)的作用下,以dNTP为原料,从引物的5′端→3′ 端延伸,合成与模板互补的DNA链。
每一循环经过变性、退火和延伸,DNA含量既增加一倍。现在有些PCR因为扩增区很短,即使Taq酶活性不是最佳也能在很短的时间内复制完成,因此可以改为两步法,即退火和延伸同时在60℃-65℃间进行,以减少一次升降温过程,提高了反应速度。
4、PCR技术的应用
PCR技术自1985年建立以来,发展之迅速,应用之广泛,表明其具有强大的生命力。近些
年来,基于PCR的基本原理,许多学者充分发挥创造性思维,对PCR技术进行研究和改进,使PCR技术得到了进上步地完善,并在此基础上派生出了许多新的用途。
反向PCR 反向PCR是用反向的互补引物来扩增两引物以外的未知序列的片段,而常规PCR扩增的是已知序列的两引物之间DNA片段。实验时选择已知序列内部没有切点的限制性内切酶对该段DNA进行酶切,然后用连接酶使带有粘性末端的靶序列环化连接,再用一对反向的引物进行PCR,其扩增产物将含有两引物外未知序列,从而对未知序进行分析研究。
不对称PCR 不对称PCR是用不等量的一对引物,PCR扩增后产生大量的单链DNA(SSDNA)。这对引物分别称为非限制引物与限制性引物,其比例一般为50~100:1。在PCR反应的最初10~15个循环中,其扩增产物主要是双链DNA,但当限制性引物(低浓度引物)消耗完后,非限制性引物(高浓度引物)引导的PCR就会产生大量的单链DNA。不对称PCR的关键是控制限制性引物的绝对量,需多次摸索优化两条引物的比例。还有一种方法是先用等浓度的引物PCR扩增,制备双键DNA,(dsDNA),然后以此dsDNA为模板,再以其中的一条引物进行第二次PCR,制备ssDNA。不对称PCR制备的ssDNA,主要用于核酸序列测定。
参考文献
[1]马建岗,基因工程学原理,西安交通大学出版社,第3版
[2] 陶生策,张治平,张先恩. PCR技术研究进展[ J ]. 生物工程进展, 2001
[3] 俞华,《PCR技术发展及应用前景》,人民日报,2003.12(5),第7版
[4] 马中声等,《分子生物学》,教育出版社,北京,2004